




dopamine
(See also Overview of Movement and Cerebellar Disorders.)
The prevalence of Parkinson disease in North America is approximately 0.6% in adults age ≥ 45;prevalence increases with age (1).
The mean age at onset is approximately 6 years. Patients with tremor-dominant disease may develop motor symptoms earlier than those with akinetic-rigid disease (2).
Parkinson disease is usually idiopathic.
Juvenile parkinsonism, which is rare, begins during childhood or adolescence up to 20 years. Onset between ages 21 and 40 years is sometimes called young or early-onset Parkinson disease. Genetic causes are more likely in juvenile and early-onset Parkinson disease; these forms may differ from later-onset Parkinson disease because
They progress more slowly.
They are very sensitive to dopaminergic treatments.
Most disability results from nonmotor symptoms such as depression, anxiety, and pain.
Secondary parkinsonism is brain dysfunction that is characterized by basal ganglia dopaminergic blockade and that is similar to Parkinson disease, but it is caused by something other than Parkinson disease (eg, medications, cerebrovascular disease, trauma, postencephalitic changes).
Atypical parkinsonism refers to a group of neurodegenerative disorders that have some features similar to those of Parkinson disease but have some different clinical features, a worse prognosis, a modest or no response to levodopa, and a different pathology (eg, neurodegenerative disorders such as multiple system atrophy, progressive supranuclear palsy, dementia with Lewy bodies, and corticobasal ganglionic degeneration).
General references
1. Willis AW, Roberts E , Beck JC, et al: Incidence of Parkinson disease in North America. NPJ Parkinsons Dis 8 (1):170, 2022. doi: 10.1038/s41531-022-00410-y
2. Rajput AH, Voll A, Rajput ML, Robinson CA, Rajput A: Course in Parkinson disease subtypes: A 39-year clinicopathologic study. Neurology 73 (3):206–212, 2009. doi: 10.1212/WNL.0b013e3181ae7af1
Pathophysiology of Parkinson Disease
Synuclein is primarily a neuronal cell protein that can aggregate into insoluble fibrils and form Lewy bodies.
The pathologic hallmark of sporadic or idiopathic Parkinson disease is
Synuclein-filled Lewy bodies in the nigrostriatal system
However, synuclein can accumulate in many other parts of the nervous system, including the dorsal motor nucleus of the vagus nerve, basal nucleus of Meynert, hypothalamus, neocortex, olfactory bulb, sympathetic ganglia, and myenteric plexus of the gastrointestinal tract. Lewy bodies appear in a temporal sequence, and many experts believe that Parkinson disease is a relatively late development in a systemic synucleinopathy. Other synucleinopathies (synuclein deposition disorders) include dementia with Lewy bodies and multiple system atrophy. Parkinson disease may share features of other synucleinopathies, such as autonomic dysfunction and dementia.
Rarely, Parkinson disease occurs without Lewy bodies (eg, in a form due to a mutation in the PARK 2 gene).
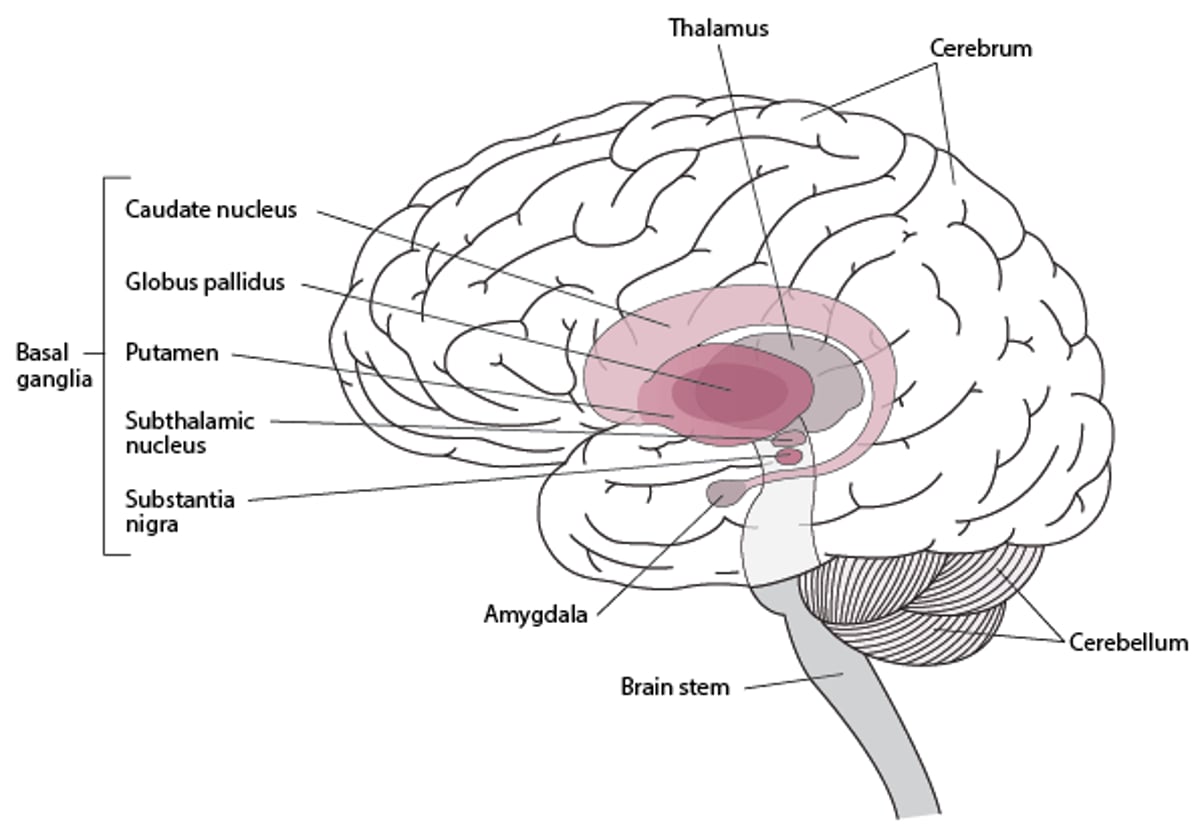
In Parkinson disease, pigmented neurons of the substantia nigra, locus ceruleus, and other brain stem dopaminergic cell groups degenerate. Loss of substantia nigra neurons results in depletion of dopamine in the dorsal aspect of the putamen (part of the basal ganglia) and causes many of the motor manifestations of Parkinson disease (see figure Basal Ganglia).
Basal Ganglia


Etiology of Parkinson Disease
Although Parkinson disease was initially considered sporadic, accumulating evidence suggests that Parkinson disease has a substantial genetic component, probably causing or influencing at least 10 to 25% of cases, more commonly in younger patients (1). The frequency of genetic mutations varies by ethnicity.
Worldwide, mutations in LRRK2 encoding for leucine-rich repeat protein kinase 2) and GBA (encoding for glucocerebrosidase), which are the two most common genetic determinants of Parkinson disease. Mutations, duplications, or triplications in the gene for alpha-synuclein are very rare, familial cases of Parkinson disease.
Etiology reference
1. Singleton AB, Farrer MJ, Bonifati V: The genetics of Parkinson’s disease: Progress and therapeutic implications. Mov Disord 28 (1):14–23, 2013. doi: 10.1002/mds.25249
Symptoms and Signs of Parkinson Disease
In most patients, symptoms of Parkinson disease begin insidiously.
A resting tremor of one hand is often the first symptom. The tremor is characterized as follows:
Slow and coarse
Maximal at rest, lessening during movement, and absent during sleep
Amplitude increased by emotional tension or fatigue
Often involving the wrist and fingers, sometimes involving the thumb moving against the index finger (pill rolling), as when people roll a pill in their hand or handle a small object
Usually, the hands or feet are affected first, most often asymmetrically. The jaw and tongue may also be affected, but not the voice. However, speech may become hypophonic, with characteristic monotonous, sometimes stuttering dysarthria. Tremor may become less prominent as rigidity progresses. In predominantly rigid-akinetic forms of Parkinson disease, resting tremor is subtle or absent.
Rigidity develops independently of tremor in many patients. When a clinician moves a rigid joint, semirhythmic jerks occur because the intensity of the rigidity varies, causing a ratchet-like effect (cogwheel rigidity).
Slow movements (bradykinesia) are typical in Parkinson disease. Repetitive motor activity results in a progressive or sustained decrease in amplitude of movement (hypokinesia), and movement becomes hard to initiate (akinesia).
Rigidity and hypokinesia may contribute to muscle aches and sensations of fatigue. The face becomes masklike (hypomimic), with an open mouth and reduced blinking. Excessive drooling (sialorrhea) may contribute to disability.
Hypokinesia and impaired control of distal muscles cause micrographia (writing in very small letters) and make activities of daily living increasingly difficult.
Postural instability may develop later in Parkinson disease; if present at disease onset, alternative diagnoses should be suspected. Patients have difficulty starting to walk, turning, and stopping. They shuffle, taking short steps, holding their arms flexed to the waist, and swinging their arms little or not at all with each stride. Steps may inadvertently quicken, while stride length progressively shortens; this gait abnormality, called festination, is often a precursor to freezing of gait (when, without warning, walking and other voluntary movements may suddenly halt). A tendency to fall forward (propulsion) or backward (retropulsion) when the center of gravity is displaced results from loss of postural reflexes. Posture becomes stooped.
Dementia develops in about one third of patients, usually late in Parkinson disease. Early predictors of its development are visuospatial impairment (eg, getting lost while driving) and decreased verbal fluency.
Sleep disorders are common. Insomnia may result from nocturia or from the inability to turn in bed. Sleep deprivation may exacerbate depression and cognitive impairment and contribute to excessive daytime sleepiness. Rapid eye movement (REM) sleep behavior disorder may develop; in this disorder, verbalization and uncontrollable, possibly violent limb movements occur during REM sleep because the paralysis that normally occurs during REM sleep is absent. REM sleep behavior disorder is often accompanied by early neurodegenerative signs that occur primarily in patients with alpha-synucleinopathies, which can precede and/or increase the risk of developing Parkinson disease, multiple system atrophy, or dementia with Lewy bodies.
Neurologic symptoms unrelated to parkinsonism commonly develop because synucleinopathy occurs in other areas of the central, peripheral, and autonomic nervous systems. The following are examples:
Almost universal sympathetic denervation of the heart, contributing to orthostatic hypotension
Esophageal dysmotility, contributing to dysphagia and increased risk of aspiration
Lower bowel dysmotility, contributing to constipation
Urinary hesitancy and/or urgency, potentially leading to incontinence (common)
Anosmia (common)
In some patients, some of these symptoms occur before the motor symptoms of Parkinson disease and frequently worsen over time.
Seborrheic dermatitis is also common.
Diagnosis of Parkinson Disease
Mainly clinical evaluation, based on motor symptoms
Diagnosis of Parkinson disease is clinical. Parkinson disease is suspected in patients with characteristic unilateral resting tremor, decreased movement, or rigidity. During finger-to-nose coordination testing, the tremor disappears (or attenuates) in the limb being tested.
During the neurologic examination, patients cannot perform rapidly alternating or rapid successive movements well. Sensation and strength are usually normal. Reflexes are normal but may be difficult to elicit because of marked tremor or rigidity.
Slowed and decreased movement due to Parkinson disease must be differentiated from decreased movement and spasticity due to lesions of the corticospinal tracts. Unlike Parkinson disease, corticospinal tract lesions cause
Paresis (weakness or paralysis), preferentially in distal antigravity muscles
Hyperreflexia
Extensor plantar responses (Babinski sign)
Spasticity that increases muscle tone in proportion to the rate and degree of stretch placed on a muscle until resistance suddenly melts away (clasp-knife phenomenon)
The diagnosis of Parkinson disease is supported by the presence of other signs such as infrequent blinking, lack of facial expression, and gait abnormalities. Postural instability is also present, but if it occurs early in the disease, clinicians should consider other possible diagnoses.
In older adults, other possible causes of decreased spontaneous movements or a short-stepped gait, such as severe depression, hypothyroidism, or use of antipsychotics or certain antiemetics, must be excluded before Parkinson disease is diagnosed.
levodopa
Causes of secondary or atypical parkinsonism can be identified by
A thorough history, including occupational, medication, and family history
Evaluation for neurologic deficits characteristic of disorders other than Parkinson disease
Neuroimaging when patients have atypical features (eg, early falls, early cognitive impairment, ideomotor apraxia [inability to imitate hand gestures], hyperreflexia)
Treatment of Parkinson Disease
Dopamine agonists
Catechol Olevodopa is wearing off
Surgery if medications do not sufficiently control symptoms or have intolerable adverse effects
Exercise and adaptive measures
Many oral medications are commonly used to relieve symptoms of Parkinson disease ([1]; see table Some Commonly Used Oral Antiparkinsonian Medications).
is the most effective treatment. However, when Parkinson disease becomes severe, sometimes soon after diagnosis, response to levodopa can wear off, causing fluctuations in motor symptoms and dyskinesias (see below). To reduce the time levodopa is taken and thus minimize these effects, clinicians can consider treating younger patients who have mild disability with the following medications first:
Dopamine
levodopa becomes ineffective because of disease progression rather than cumulative exposure to levodopa, as was previously believed, so early use of levodopa probably does not hasten its ineffectiveness.
Doses are often reduced in older adults. Medications that cause or worsen symptoms, particularly antipsychotics, are avoided.
Levodopa
dopamine, crosses the blood-brain barrier into the basal ganglia, where it is decarboxylated to form dopaminelevodopa from being decarboxylated into dopamine outside the brain (peripherally), thus lowering the levodopa dosage required to produce therapeutic levels in the brain and minimizing adverse effects due to dopamine in the peripheral circulation.
Levodopa is most effective at relieving bradykinesia and rigidity and often substantially reduces tremor (2).
Common short-term adverse effects of levodopa are
Nausea
Vomiting
Light-headedness
Common long-term adverse effects include
Mental and psychiatric abnormalities (eg, delirium with confusion, psychoses, paranoia, visual hallucinations, punding [complex, repetitive, stereotyped behaviors])
Motor dysfunction (eg, dyskinesias, motor fluctuations)
Hallucinations and paranoia occur most often in older adults and in patients who have cognitive impairment or dementia.
The dose that causes dyskinesias tends to decrease as the disease progresses. Over time, the dose that is needed for therapeutic benefit and the one that causes dyskinesia converge.
Preferably, levodopa should not be given with food because protein can reduce absorption of levodopa. Four to 5 doses of levodopa a day are recommended to decrease the effect of fluctuating plasma levels of levodopa on different basal ganglia, which, can cause motor fluctuations and dyskinesias.
If peripheral adverse effects of levodopa (eg, nausea, vomiting, postural light-headedness) predominate, increasing the amount of carbidopa may help. Carbidopa doses up to 150 mg are safe and do not decrease the efficacy of levodopa.
Domperidone can be used to treat the adverse effects of levodopa (and other antiparkinsonian medications). It blocks peripheral dopamine receptors and does not cross the blood-brain barrier to affect the brain. By decreasing the decarboxylation of levodopa to dopamine, domperidone lessens the peripheral adverse effects of levodopa, thereby decreasing nausea, vomiting, and orthostatic hypotension. The recommended dosages are
Immediate-release: 10 mg orally 3 times a day, increased up to 20 mg 3 times a day if needed
Sustained-release: 30 to 60 mg once in the morning (this dose may be enough to control levodopa's peripheral adverse effects)
Domperidone is not routinely available in the United States.
carbidopa/levodopa.
Occasionally, levodopa must be used to maintain motor function despite levodopa-induced hallucinations or delirium. In such cases, hallucinations and delirium can be treated with medications.
Psychosis3
After 2 to 5 years of treatment, most patients experience fluctuations in their response to levodopa, and symptom control may fluctuate unpredictably between effective and ineffective (on-off fluctuations), as response to levodopa starts to wear off. Symptoms may occur before the next scheduled dose (called off effects). The dyskinesias and off effects result from a combination of the pharmacokinetic properties of levodopa (particularly its short half-life as an oral medication), and disease progression.
Early in Parkinson disease, there are enough surviving neurons to buffer any oversaturation of dopaminergic receptors in the substantia nigra. As a result, dyskinesias are less likely to occur, and levodopa's therapeutic effect lasts longer because of the reuptake of excessive levodopa and its reutilization. As dopaminergic neurons are further depleted, each dose of levodopa saturates more and more dopamine receptors, resulting in dyskinesias and motor fluctuations because the delivery of levodopa to the substantia nigra becomes dependent on the plasma half life of levodopa (1.5 to 2 hours).
However, dyskinesias result mainly from disease progression and are not directly related to cumulative exposure to levodopa, as previously believed. Disease progression is associated with pulsatile administration of oral levodopa, which sensitizes and changes glutamatergic receptors, especially NMDA (N-methyl-d-aspartate) receptors. Eventually, the period of improvement after each dose shortens, and drug-induced dyskinesias result in swings from akinesia to dyskinesias. Traditionally, such swings are managed by keeping the levodopa dose as low as possible and using dosing intervals as short as every 1 to 2 hours, which are impractical. Alternative methods to decrease the off (akinetic) times include adjunctive use of dopamine
Amantadine
Lessen tremors
levodopa’s effects. It may augment dopaminergic activity, anticholinergic effects, or both. Amantadine is also an NMDA-receptor antagonist and thus may help slow the progression of Parkinson disease and dyskinesias. If used as monotherapy, amantadine often loses its effectiveness after several months.
Dopamine agonists
These medications directly activate dopamine receptors in the basal ganglia. They include
dopamine agonists may be useful at all stages of the disease, including as adjunctive therapy in later stages. Adverse effects may limit the use of oral dopamine agonists. In 1 to 2% of patients, these medications may cause compulsive gambling, excessive shopping, hypersexuality, or overeating, requiring dose reduction or withdrawal of the causative medication and possibly avoidance of the medication class.
and
, given transdermally once a day, provides more continuous dopaminergic stimulation than medications given via other routes. Dose starts at 2 mg once a day and is usually increased to 6 mg once a day. Outside the United States, higher doses (8 mg) may be recommended.
is adopamineorthostatic hypotension. Blood pressure (BP) is checked in the supine and standing positions before treatment and 20, 40, and 60 minutes afterward. Other adverse effects are similar to those of other dopamineapomorphine and continuing trimethobenzamide for the first 2 months of treatment.
Apomorphine given by subcutaneous pump is available in some countries; it can be used instead of a levodopa pump in patients who have advanced Parkinson disease and who are not candidates for functional surgery.
Pergolide, an older ergot-derived dopamine agonist, was taken off the market because it increased the risk of cardiac valve fibrosis.
Selective MAO-B inhibitors
inhibits one of the two major enzymes that break down dopamineselegiline helps prolong levodopa’s effectiveness. Used initially as monotherapy, selegilineamphetamine-like metabolites, is sometimes triggered when patients taking a nonselective MAO inhibitor consume tyramine in foods (eg, some cheeses). Although virtually free of adverse effects, selegiline can potentiate levodopa-induced dyskinesias, mental and psychiatric adverse effects, and nausea, requiring reduction in the levodopaselegiline).
selegiline. Unlike selegiline, it does not have amphetamine-like metabolites, so theoretically, risk of a hypertensive crisis when patients consume tyramine is lower with rasagiline.
Anticholinergic medications
Some studies using a mouse model indicate that use of anticholinergic medications should be limited because they appear to increase tau pathology and neurodegeneration; degree of increase correlates with the medication's central anticholinergic activity (4, 5).
Commonly used anticholinergic medications include
levodopa.
Catechol O-methyltransferase (COMT) inhibitors
levodopa and dopaminelevodopa for a long time when response to levodopa is progressively wearing off at the end of dosing intervals (known as wearing-off effects),
levodopa
Tolcapone should be stopped if alanine aminotransferase (ALT) or aspartate aminotransferase (AST) levels increase to twice the upper limit of the normal range or higher or if symptoms and signs suggest that the liver is damaged.
opicapone does not require monitoring with periodic laboratory tests or multiple oral dosing. Recommended dose is 50 mg at bedtime.
Surgery
If pharmacotherapy is ineffective and/or has intolerable adverse effects, surgery, including deep brain stimulation and lesional surgery, may be considered.
Deep brain stimulation of the subthalamic nucleus or globus pallidus interna is often recommended for patients with levodopa-induced dyskinesias or significant motor fluctuations; this procedure can modulate overactivity in the basal ganglia and thus decrease parkinsonian symptoms in patients with Parkinson disease. For patients with tremor only, stimulation of the ventralis intermediate nucleus of the thalamus is sometimes recommended; however, because most patients also have other symptoms, stimulation of the subthalamic nucleus, which relieves tremor as well as other symptoms, is usually preferred. When the main problem is inadequate control of dyskinesias or when patients have an increased risk of cognitive decline, the globus pallidus interna is a good target.
Lesional surgery aims to stop overactivity directed to the thalamus from the globus pallidus interna; thalamotomy is sometimes done to control tremor in patients with tremor-predominant Parkinson disease. However, lesional surgery is not reversible and cannot be modulated over time; bilateral lesional surgery is not recommended because it can have severe adverse effects such as dysphagia and dysarthria. Lesional surgery involving the subthalamic nucleus is contraindicated because it causes severe ballismus.
Patient selection is the most important factor for successful functional surgery in Parkinson disease. Surgery is usually considered when pharmacotherapy of dyskinesias and/or motor fluctuations is ineffective or severely limited. Pharmacotherapy may be inadequate because the medication has adverse effects that prevent further increases in the levodopa dose, which might lessen symptoms.
Other selection criteria include
Parkinson disease present for 5 to 15 years
Patient age < 70 years
No significant cognitive decline, no affective disorder, and, depending on life expectancy, no terminal disease (eg, cancer, chronic renal failure, liver failure, significant cardiopathy, uncontrolled diabetes mellitus or hypertension)
Patients with cognitive impairment, dementia, or a psychiatric disorder are not suitable candidates for surgery because neurosurgery can exacerbate cognitive impairment and psychiatric disorders, and the risk of additional mental impairment outweighs the benefits of any improvement in motor function.
High-intensity focused ultrasound (HIFU)
MR-guided high-intensity focused ultrasound can be used to control severe tremor refractory to medications in patients with Parkinson disease. With this procedure, the ventral intermediate nucleus of the thalamus can be ablated with minimal risk of hemorrhage and infection, which may occur when more invasive neurosurgical procedures are used.
In addition to tremor, HIFU has shown promise in treating symptoms of bradykinesia and rigidity while reducing the rate of dyskinesias (6). Although HIFU being less invasive than other surgical interventions, deep brain stimulation is favored by most expert clinicians.
Physical measures
Maximizing activity is a goal. Patients should increase daily activities to the greatest extent possible. If they cannot, physical or occupational therapy, which may involve a regular exercise program, may help condition them physically. Therapists may teach patients adaptive strategies and help them make appropriate adaptations in the home (eg, installing grab bars to reduce the risk of falls).
Caregiver and end-of-life issues
Because Parkinson disease is progressive, patients eventually need help with normal daily activities. Caregivers should be directed to resources that can help them learn about the physical and psychologic effects of Parkinson disease and about ways to help the patient function as well as possible. Because such care is tiring and stressful, caregivers should be encouraged to contact support groups for social and psychologic support.
Eventually, most patients become severely disabled and immobile. They may be unable to eat, even with assistance. Because swallowing becomes increasingly difficult, death due to aspiration pneumonia is a risk. For some patients, a nursing home may be the best place for care.
Before people with Parkinson disease are incapacitated, they should establish advance directives, indicating what kind of medical care they want at the end of life.
Treatment references
1. Fox SH, Katzenschlager R, Lim S-Y, et al: International Parkinson and movement disorder society evidence-based medicine review: Update on treatments for the motor symptoms of Parkinson's disease. Mov Disord 33 (8):1248–1266, 2018. doi: 10.1002/mds.27372 Epub 2018 Mar 23.
2. Fahn S, Oakes D, Shoulson I, et alN Engl J Med 351 (24):2498–2508, 2004. doi: 10.1056/NEJMoa033447
3. Cummings J, Isaacson S, Mills R, et alLancet 383 (9916):533–540, 2014. doi: 10.1016/S0140-6736(13)62106-6
4. Yoshiyama Y, Kojima A, Itoh K, Uchiyama T, Arai K: Anticholinergics boost the pathological process of neurodegeneration with increased inflammation in a tauopathy mouse model. Neurobiol Dis 2012 45 (1):329–336, 2012. doi: 10.1016/j.nbd.2011.08.017
5. Yoshiyama Y, Kojima A, Itoh K, et al: Does anticholinergic activity affect neuropathology? Implication of neuroinflammation in Alzheimer's disease. Neurodegener Dis 15 (3):140-148, 2015. doi: 10.1159/000381484
6. Vibhor KrishnaV, Paul S. Fishman PS, Eisenberg HM, et al: Trial of globus pallidus focused ultrasound ablation in Parkinson's disease. N Engl J Med 388 (8):683–693, 2023. doi: 10.1056/NEJMoa2202721
Key Points
Parkinson disease is a synucleinopathy and thus can overlap with other synucleinopathies (eg, dementia with Lewy bodies, multiple system atrophy).
Suspect Parkinson disease based on characteristic features: resting tremor, muscle rigidity, slow and decreased movement, and postural and gait instability.
Distinguish Parkinson disease from disorders that cause similar symptoms based mainly on the history and physical examination results, but also test responsiveness to levodopa; sometimes neuroimaging is useful.
dopamine agonists, MAO-B inhibitors, COMT inhibitors) may be used before and/or with levodopa/carbidopa.
Consider surgical procedures, such as deep brain stimulation, if patients have symptoms refractory to optimal pharmacotherapy and do not have cognitive impairment or a psychiatric disorder.






