




Esophageal atresia is incomplete formation of the esophagus, frequently associated with tracheoesophageal fistula. The abnormality is usually suspected on prenatal ultrasound due to polyhydramnios or during the neonatal period due to aspiration pneumonia. Diagnosis is made after failure to pass a nasogastric or orogastric tube into the stomach. Treatment is surgical repair.
(See also Overview of Congenital Gastrointestinal Anomalies.)
Esophageal atresia is the most common gastrointestinal (GI) atresia. The estimated incidence is 1 in 3500 live births. Other congenital malformations are present in up to 50% of cases. Two syndromes in particular are associated with esophageal atresia:
VACTERL (vertebral anomalies, anal atresia, cardiac malformations, tracheoesophageal fistula, esophageal atresia, renal anomalies and radial aplasia, and limb anomalies)
CHARGE (coloboma, heart defects, atresia of the choanae, retardation of mental and/or physical development, genital hypoplasia, and ear abnormalities)
Approximately 19% of infants with esophageal atresia meet criteria for VACTERL.

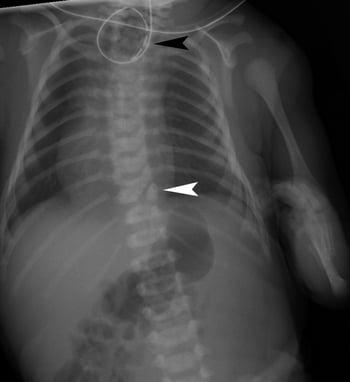
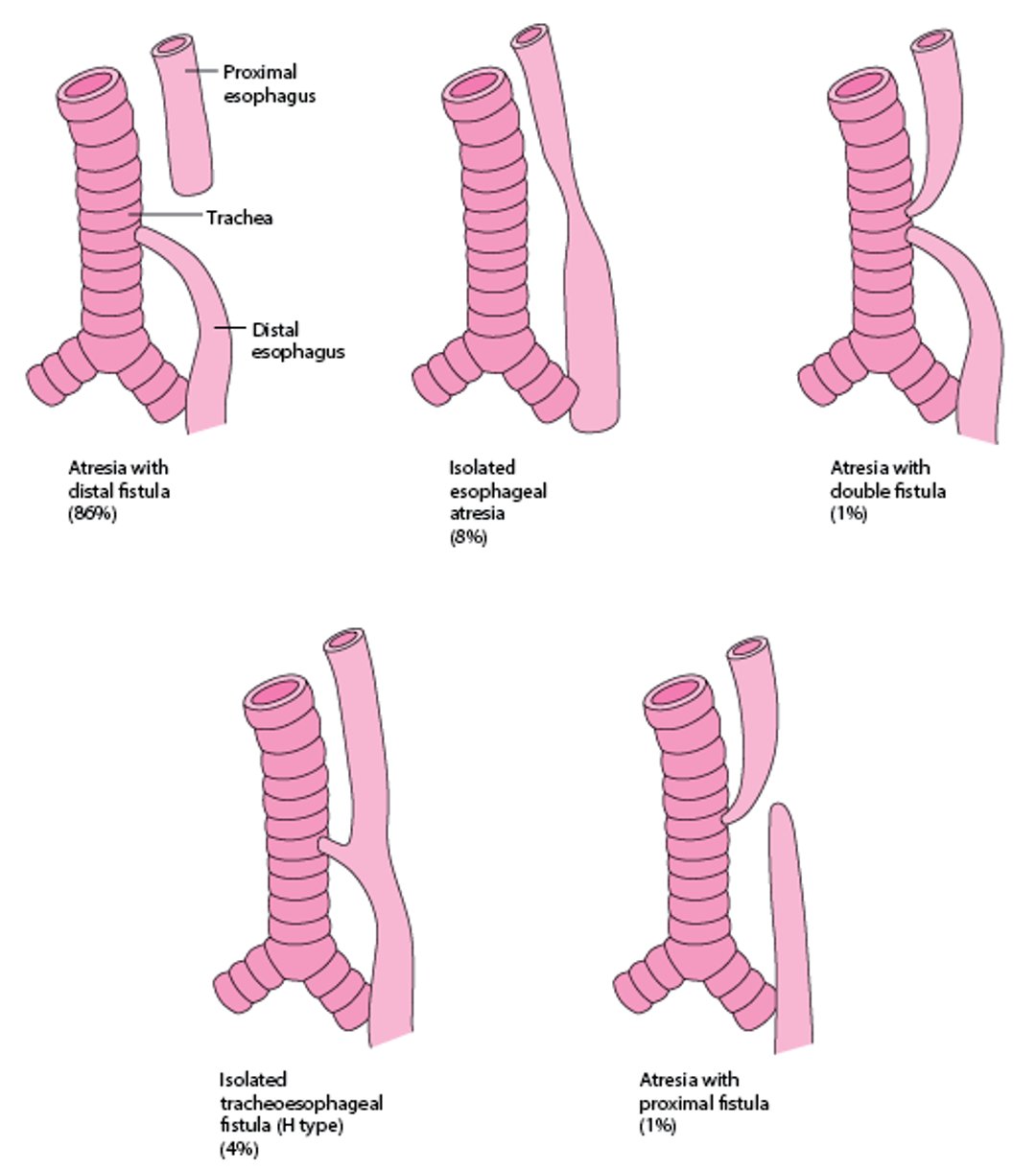
There are 5 major types of esophageal atresia (see figure Types and Relative Frequencies of Esophageal Atresia and Tracheoesophageal Fistula). Most of the types also involve a fistula between the trachea and esophagus.
Most infants present during the neonatal period, but infants with the H-type fistula may remain undiagnosed until late in infancy or childhood; there are case reports of H-type fistulas not presenting until adulthood (1).
Characteristic signs are excessive secretions, coughing and cyanosis after attempts at feeding, and aspiration pneumonia. Esophageal atresia with a distal fistula leads to abdominal distention because, as the infant cries, air from the trachea is forced through the fistula into the lower esophagus and stomach.
Types and Relative Frequencies of Esophageal Atresia and Tracheoesophageal Fistula
Relative frequencies are based on a compilation of various sources. |

General reference
1. Tiwari C, Nagdeve N, Saoji R, et al: Congenital H-type tracheo-oesophageal fistula: An institutional review of a 10-year period. J Mother Child 24(4):2-8, 2021. Published 2021 Jul 13. doi:10.34763/jmotherandchild.20202404.d-20-00004
Diagnosis of Esophageal Atresia
Prenatal: Ultrasonography
Postnatal: Nasogastric tube (NGT) or orogastric tube placement and x-ray
Routine prenatal ultrasonography may suggest esophageal atresia. Polyhydramnios may be present but is not specific to the diagnosis because it can occur with many other disorders. The fetal stomach bubble may be absent but only in fewer than half of cases. Less commonly, there is a dilated upper esophageal pouch, but this is typically looked for only in fetuses with polyhydramnios and no stomach bubble.
After delivery, an NGT or an orogastric tube is inserted if esophageal atresia is suspected by prenatal ultrasonography or clinical findings; diagnosis of esophageal atresia is suggested by inability to pass the tube into the stomach. A radiopaque catheter determines the location of the atresia on x-ray. In atypical cases, a small amount of water-soluble contrast material may be needed to define the anatomy under fluoroscopy. The contrast material should be quickly aspirated back because it can cause a chemical pneumonitis if it enters the lungs. This procedure should be done only by an experienced radiologist at the center where neonatal surgery will be done.
Treatment of Esophageal Atresia
Surgical repair
Preoperative management aims to get the infant into optimal condition for surgery and prevent aspiration pneumonia, which makes surgical correction more hazardous. Oral feedings are withheld. Continuous suction with an NGT in the upper esophageal pouch prevents aspiration of swallowed saliva. The infant should be positioned prone with the head elevated 30 to 40° and with the right side down to facilitate gastric emptying and minimize the risk of aspirating gastric acid through the fistula. If definitive repair must be deferred because of extreme prematurity, aspiration pneumonia, or other congenital malformations, a gastrostomy tube is placed to decompress the stomach. Suction through the gastrostomy tube then reduces the risk that gastric contents will reflux through the fistula into the tracheobronchial tree.
Surgical repair
When the infant’s condition is stable, extrapleural surgical repair of the esophageal atresia and closure of the tracheoesophageal fistula can be done (1). If a fistula is noted, it needs to be ligated. In about 90% of cases, primary anastomosis of the esophagus can be done. In the remaining cases, where an extremely long gap exists, options are to do a gastric transposition procedure or a colonic interposition procedure.
Some pediatric surgeons do a Foker procedure. In this procedure, traction sutures are placed in the ends of the esophageal pouches, brought out through the skin, and fixed with silastic buttons. Traction is gradually applied to the sutures, which stimulates elongation of the esophagus by as much as 1 to 2 mm/day. Once the ends of the esophagus have come together, or are in close proximity, a primary anastomosis is done (2).
The most common acute complications are leakage at the anastomosis site and stricture formation. Feeding difficulties are common after successful surgical repair because of poor motility of the distal esophageal segment, which occurs in up to 85% of cases. This poor motility predisposes the infant to gastroesophageal reflux, which occurs in up to 50% of children, as well as dysphagia. It is recommended that all neonates with esophageal atresia be treated for reflux with acid-suppressive therapy (3). If medical management for reflux fails, a Nissen fundoplication may be required.
Treatment references
1. Dingemann C, Eaton S, Aksnes G, et al: ERNICA Consensus Conference on the Management of Patients with Esophageal Atresia and Tracheoesophageal Fistula: Diagnostics, Preoperative, Operative, and Postoperative Management. Eur J Pediatr Surg 30(4):326-336, 2020. doi:10.1055/s-0039-1693116
2. Shieh HF, Jennings RW: Long-gap esophageal atresia. Semin Pediatr Surg 26(2):72–77, 2017. doi: 10.1053/j.sempedsurg.2017.02.009
3. Krishnan U, Mousa H, Dall’Oglio L, et al: ESPGHAN-NASPGHAN guidelines for the evaluation and treatment of gastrointestinal and nutritional complications in children with esophageal atresia-tracheoesophageal fistula. J Pediatr Gastroenterol Nutr 63(5):550–570, 2016. doi: 10.1097/MPG.0000000000001401
Key Points
There are 5 types of esophageal atresia; all but one also involve a tracheoesophageal fistula.
Sometimes diagnosis is suspected based on prenatal ultrasonography.
Clinical manifestations include excessive secretions, coughing, and cyanosis after attempts at feeding, and aspiration pneumonia; an H-type fistula usually presents later in life and is suspected because of aspiration pneumonia.
Diagnose by passing a nasogastric or an orogastric tube and x-ray.
Treat with surgical repair and acid-suppressive therapy.






