




Sturge-Weber syndrome is a congenital vascular disorder characterized by a facial port-wine nevus, a leptomeningeal angioma, and neurologic complications (eg, seizures, focal neurologic deficits, intellectual disability). Diagnosis is clinical. Treatment is symptomatic.
Sturge-Weber syndrome is a neurocutaneous syndrome that occurs in 1 in 20,000 people (1). Sturge-Weber syndrome is not inherited. It is caused by a somatic mutation (a change in DNA that occurs after conception in the precursors of the affected area) in the GNAQ gene on chromosome 9q21.
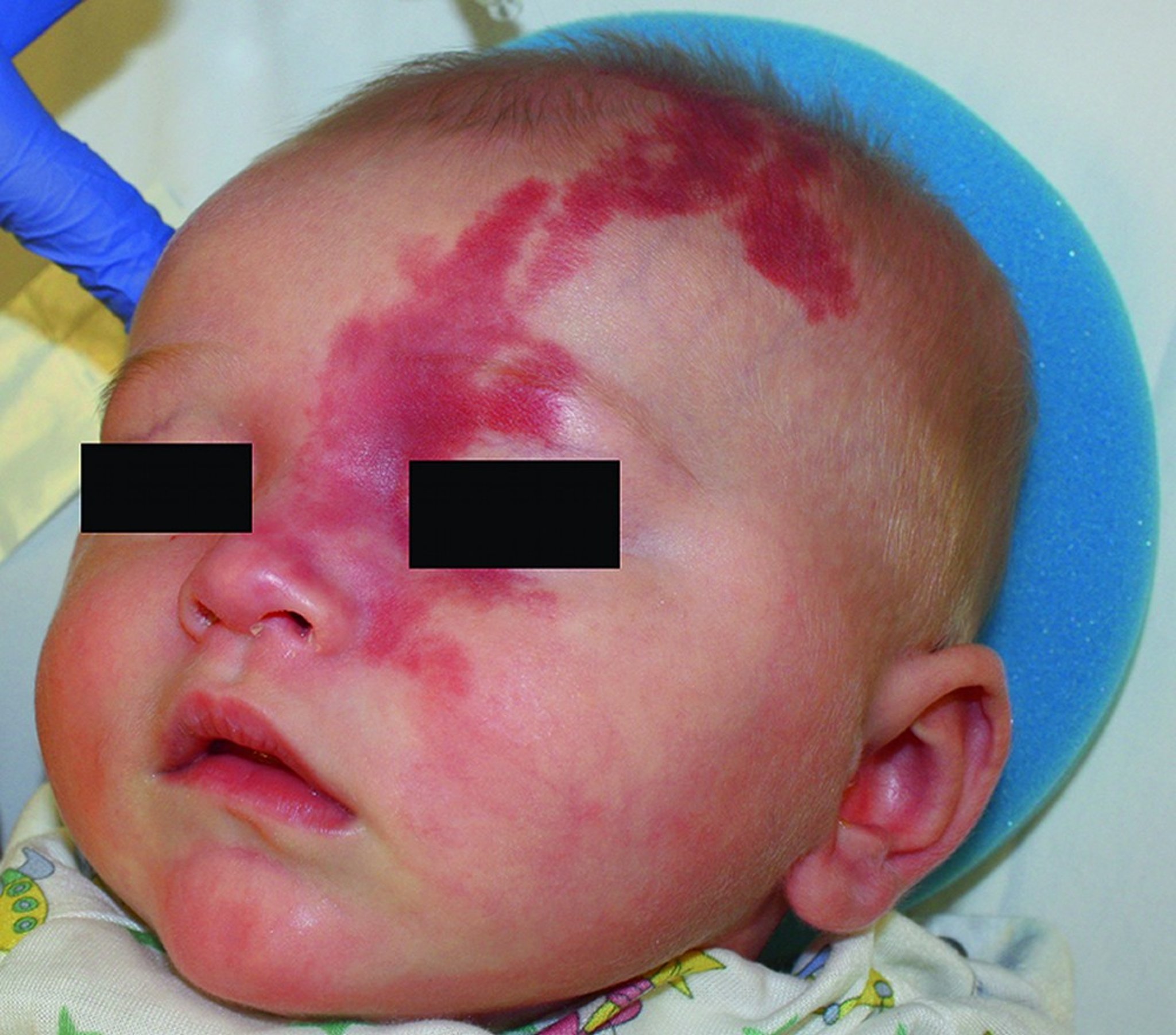
Sturge-Weber syndrome causes a capillary malformation called a port-wine nevus (or sometimes a stain or birth mark) typically on the forehead and upper eyelid in the distribution of the 1st and/or 2nd division of the trigeminal nerve. A similar vascular lesion—leptomeningeal angioma—occurs in 90% of patients when the port-wine nevus involves upper and lower eyelids on one side but occurs in only 10 to 20% of patients when only one eyelid is affected. Usually, the nevi and leptomeningeal angiomas are unilateral, but, rarely, patients have bilateral port-wine nevi in the distribution of the 1st division of the trigeminal nerve and bilateral leptomeningeal angiomas.
A port-wine nevus may occur without a leptomeningeal angioma and its accompanying neurologic signs; in such cases, the eyes and eyelids may or may not be involved. Rarely, a leptomeningeal angioma occurs without the port-wine nevus and ocular involvement.

© Springer Science+Business Media
Neurologic complications of Sturge-Weber syndrome include seizures, focal neurologic deficits (eg, hemiparesis), and intellectual disability.
Sturge-Weber syndrome can also cause glaucoma and vascular narrowing, which may increase risk of stroke due to thrombosis, venous occlusion, or infarction.
Often, the involved cerebral hemisphere progressively atrophies.
Reference
1. Comi AM: Sturge-Weber syndrome. Handb Clin Neurol 132:157-168, 2015. doi: 10.1016/B978-0-444-62702-5.00011-1
Symptoms and Signs of Sturge-Weber Syndrome
The port-wine nevus can vary in size and color, ranging from light pink to deep purple.
Seizures occur in about 75 to 90% of patients and typically start by age 1 year. Seizures are usually focal but can become generalized. Hemiparesis of the side opposite the port-wine nevus occurs in 25 to 50% of patients. Sometimes the hemiparesis worsens, especially in patients whose seizures cannot be controlled.
About 50% of patients have intellectual disability, and more have some kind of learning difficulty. Development may be delayed.
Glaucoma may be present at birth or develop later. The eyeball may enlarge and bulge out of its socket (buphthalmos).
Diagnosis of Sturge-Weber Syndrome
Brain MRI or head CT
Diagnosis of Sturge-Weber syndrome is suggested by a characteristic port-wine nevus.
Brain MRI with contrast is used to check for a leptomeningeal angioma, but the angioma may not yet show up in very young children. If MRI is not available, head CT may be done; it may show calcifications in the cortex under the leptomeningeal angioma. The parallel curvilinear railroad-track calcifications seen on skull x-rays as mentioned in older literature develop during adulthood.
A neurologic examination is done to check for neurologic complications, and an ophthalmologic examination is done to check for eye complications.
Treatment of Sturge-Weber Syndrome
Symptomatic treatment
Treatment of Sturge-Weber syndrome is focused on symptoms.
Antiseizure medications and medications to treat glaucoma are used. Sometimes hemispherectomy is done if patients have intractable seizures.
Selective photothermolysis (pulsed-dye laser) can lighten the port-wine nevus.
Key Points
Sturge-Weber syndrome is a congenital vascular disorder characterized by a facial port-wine nevus, a leptomeningeal angioma, and neurologic complications.
Seizures occur in about 75 to 90% of patients and typically start by age 1 year.
About 50% of patients have intellectual disability, and more have some kind of learning difficulty.
Diagnose by noting the characteristic port-wine nevus and performing brain imaging to check for a leptomeningeal angioma.






