


Polycystic kidney disease is a hereditary disorder in which many fluid-filled sacs (cysts) form in both kidneys. The kidneys grow larger but have less functioning tissue.
Polycystic kidney disease is caused by an inherited gene defect.
Some people have such mild symptoms that they do not realize they have a disorder, but others have pain in the side, blood in the urine, high blood pressure, and crampy pain caused by kidney stones.
Diagnosis is based on ultrasonography, computed tomography, or magnetic resonance imaging of the kidneys.
The kidney stones and infections are treated, but more than half of affected people eventually need dialysis or kidney transplantation.
There are several genetic defects that cause polycystic kidney disease (PKD). Several types are caused by dominant genes, and one rare type is caused by a recessive gene. That is, a person with the disease has inherited either one copy of a dominant gene from one parent or two copies of a recessive gene, one from each parent. People with dominant gene inheritance usually have no symptoms until adulthood. People with recessive gene inheritance develop severe illness in childhood.
Polycystic Kidney Disease
In polycystic kidney disease, many cysts form in both kidneys. The cysts gradually enlarge, destroying some or most of the normal tissue in the kidneys. |

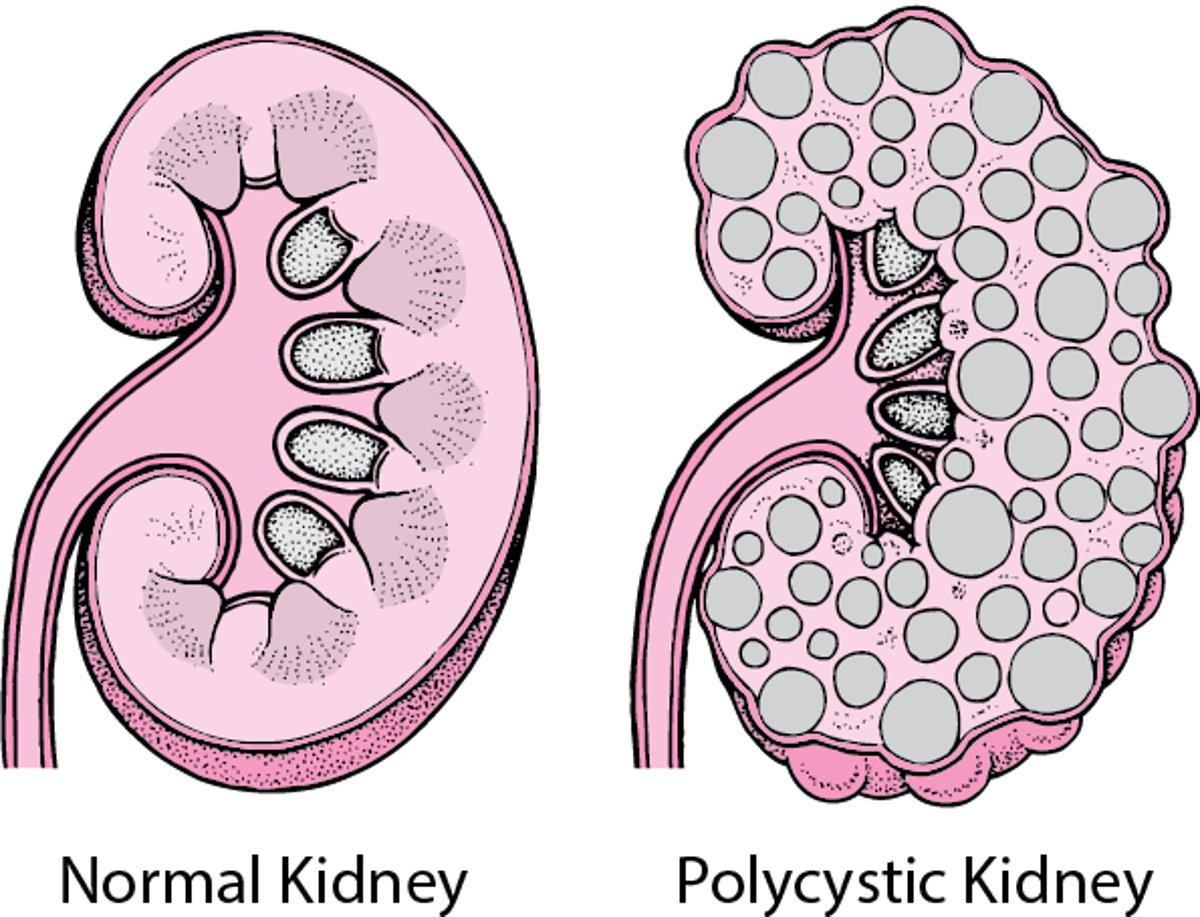
The genetic defect leads to the widespread formation of cysts in the kidneys. Gradual enlargement of the cysts with increasing age is accompanied by a reduction of blood flow and scarring within the kidneys. Cysts may become infected or bleed. Kidney stones may develop. Chronic kidney disease can occur eventually. The genetic defect may also cause cysts to develop in other parts of the body, such as the liver and pancreas.
Symptoms of Polycystic Kidney Disease
In the rare, recessive form of PKD that begins during childhood, the cysts become very large and cause the abdomen to protrude. A severely affected newborn may die shortly after birth because kidney failure can develop in the fetus, leading to poor development of the lungs. The liver is also affected, and at 5 to 10 years of age, a child with this disorder tends to develop portal hypertension, or high pressure in the blood vessels that connect the intestine and the liver (portal system). Eventually, liver failure and chronic kidney disease occur.
In the more common, dominant form of PKD, the cysts develop slowly in number and size. Typically, symptoms begin in early or middle adulthood, most often while people are in their 20s. Sometimes symptoms are so mild that people with the disease will live their whole life without ever having known they had the disorder.
Symptoms usually include discomfort or pain in the side (flank) or abdomen, blood in the urine, frequent urination, and intense crampy (colicky) pain from kidney stones. In other cases, fatigue, nausea, and other consequences of chronic kidney disease may result because the person has less functioning kidney tissue. Sometimes cysts may rupture, causing a fever that may last for weeks. Repeated urinary tract infections can worsen chronic kidney disease. At least half of people with PKD have high blood pressure by the time the disorder is recognized.
Complications
About one third of people who have the dominant form of PKD also have cysts in their liver, but these cysts do not affect liver function. People may also develop abdominal hernias or outpouchings of the wall of the large intestine (diverticulosis) and have disorders of heart valves. As many as 10% of people have dilated blood vessels (aneurysms) in their brain. Many of these brain aneurysms bleed and cause strokes.
Diagnosis of Polycystic Kidney Disease
Imaging tests
Genetic testing
A doctor suspects PKD on the basis of family history or if an imaging test done for another reason shows enlarged kidneys and cysts in the kidneys. Ultrasonography and computed tomography (CT) or magnetic resonance imaging (MRI) reveal the characteristic appearance of cysts in the kidneys and liver. Even if the person has a family history of PKD, doctors may not recommend diagnostic tests until symptoms occur because there are no effective treatments until symptoms occur and being diagnosed may have harmful effects (for example, difficulty obtaining life insurance).
Genetic testing is available to help people with PKD understand the probability that their children will inherit the condition.
Treatment of Polycystic Kidney Disease
Treatment of complications and symptoms
Effective treatment of urinary tract infections and high blood pressure slows the rate of kidney destruction. Usually angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are used to control blood pressure. However, more than half of people who have this disease develop chronic kidney disease with end-stage kidney disease (end-stage kidney failure or end-stage renal disease) at some time in their life and require dialysis or kidney transplantation.
If cysts cause severe pain, doctors may try to drain fluid from the cyst (aspiration). Aspiration may relieve pain but does not affect the person's long-term prognosis. If symptoms are very severe, the kidney may need to be removed.
More Information
The following English-language resources may be useful. Please note that THE MANUAL is not responsible for the content of these resources.
American Kidney Fund (AKF): Information about kidney disease, kidney transplant, and needs-based financial assistance to help manage medical expenses
National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK): General information on kidney diseases, including research discoveries, statistics, and community health and outreach programs
National Kidney Foundation (NKF): Information on everything from the basics of kidney function to access to treatment and support for people with kidney disease






