


(See also Overview of Nephritic Syndrome.)
IgA nephropathy is a nephritic syndrome, a form of chronic glomerulonephritis characterized by the deposition of IgA immune complexes in glomeruli. It is the most common form of glomerulonephritis worldwide. It occurs at all ages, with a peak onset in the teens and 20s; affects men 2 to 6 times more frequently than women; and is more common in White and Asian people than in Black people. Prevalence estimates for IgA kidney deposits are 5% in the United States, 10 to 20% in southern Europe and Australia, and 30 to 40% in Asia. However, some people with IgA deposits do not develop clinical disease.
Cause is unknown, but evidence suggests that there may be several mechanisms, including
Increased IgA1 production
Defective IgA1 glycosylation causing increased binding to mesangial cells
Decreased IgA1 clearance
A defective mucosal immune system
Overproduction of cytokines stimulating mesangial cell proliferation
Familial clustering has also been observed, suggesting genetic factors at least in some cases.
Renal function is initially normal, but symptomatic renal disease may develop. A few patients present with acute kidney injury or chronic kidney disease, severe hypertension, or nephrotic syndrome.
Symptoms and Signs of IgA Nephropathy
The most common manifestations are persistent or recurrent macroscopic hematuria or asymptomatic microscopic hematuria with mild proteinuria. Flank pain and low-grade fever may accompany acute episodes. Other symptoms are usually not prominent.
Gross hematuria usually begins 1 or 2 days after a febrile mucosal (upper respiratory, sinus, enteral) illness, thus mimicking acute postinfectious glomerulonephritis, except the onset of hematuria is earlier (coinciding with or immediately after the febrile illness). When this occurs with an upper respiratory illness, it is sometimes referred to as synpharyngitic hematuria.
Rapidly progressive glomerulonephritis is the initial manifestation in < 10% of patients.
Diagnosis of IgA Nephropathy
Urinalysis
Renal biopsy
Diagnosis is suggested by any of the following:
Gross hematuria, particularly within 2 days of a febrile mucosal illness or with flank pain
Incidentally noted findings on urinalysis
Occasionally, rapidly progressive glomerulonephritis
When manifestations are moderate or severe, diagnosis is confirmed by biopsy.
Urinalysis demonstrates microscopic hematuria, usually with dysmorphic red blood cells (RBCs) and occasionally RBC casts. Mild proteinuria (< 1 g/day) is typical and may occur without hematuria; nephrotic syndrome develops in ≤ 20%. Serum creatinine level is usually normal.

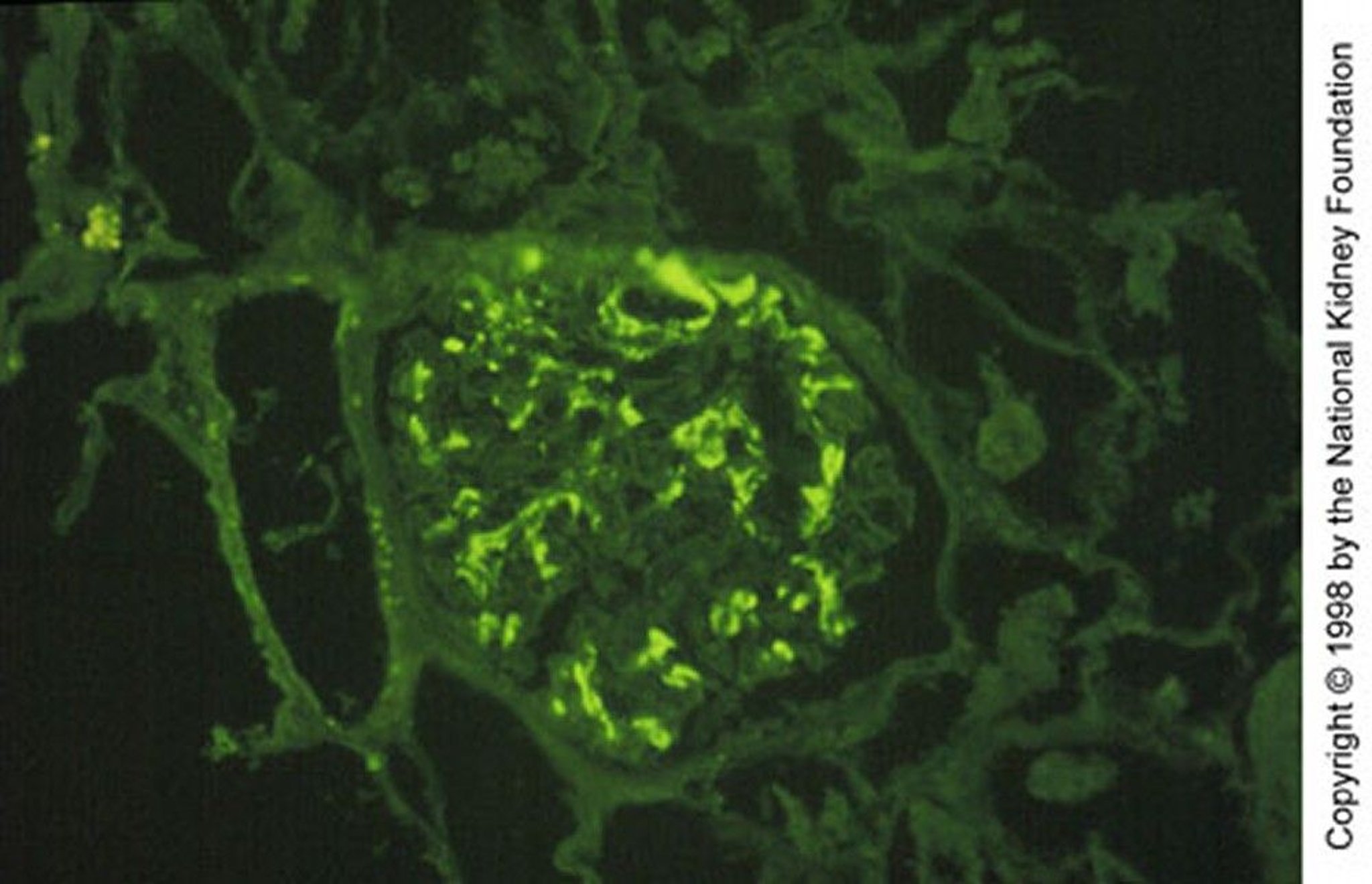
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).

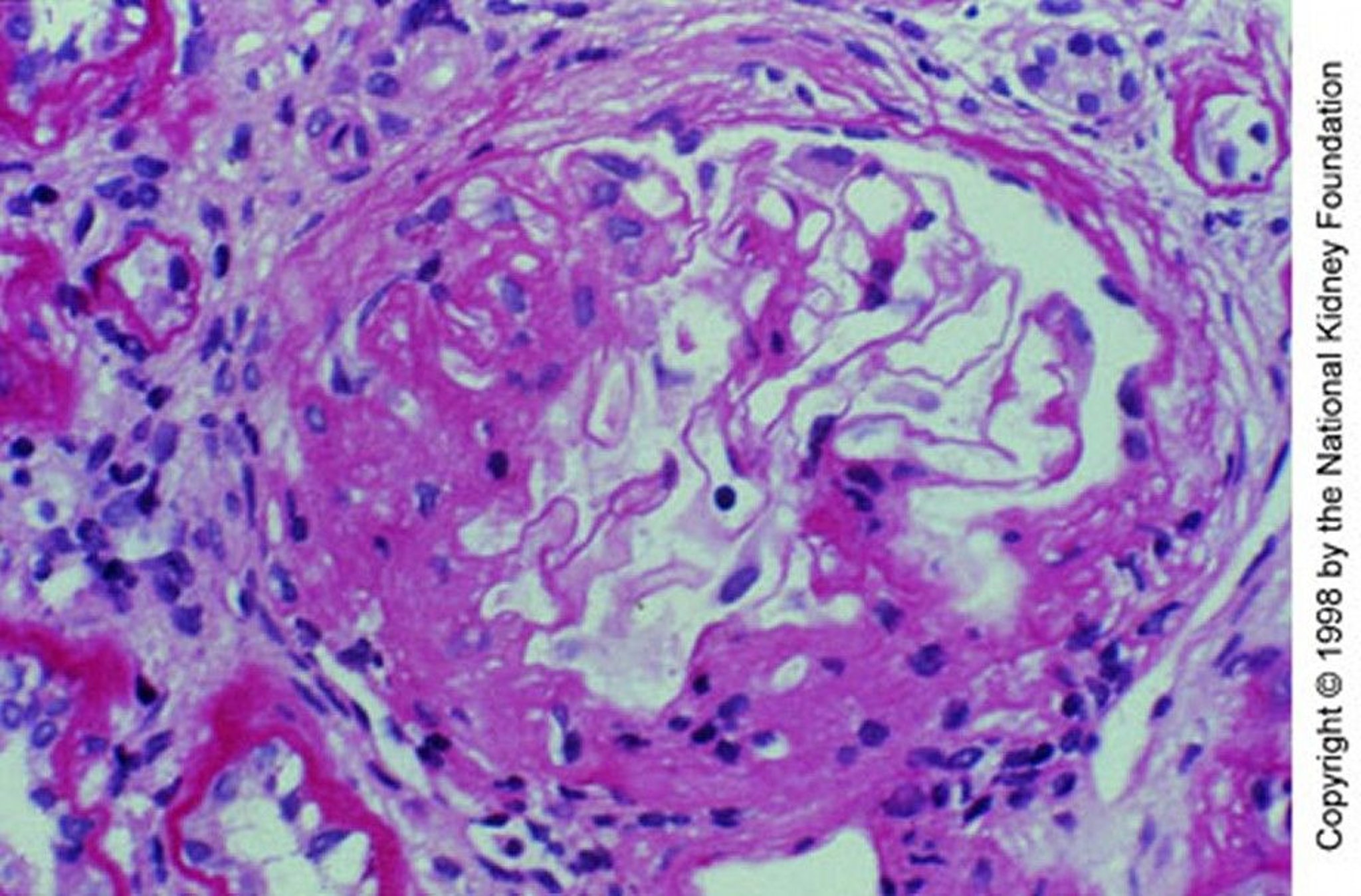
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Renal biopsy shows granular deposition of IgA and complement (C3) on immunofluorescent staining in an expanded mesangium with foci of segmental proliferative or necrotizing lesions. Importantly, mesangial IgA deposits are nonspecific and also occur in many other disorders, including immunoglobulin A vasculitis, cirrhosis, inflammatory bowel disease, celiac disease, psoriasis, HIV infection, lung cancer, and several systemic rheumatic diseases.
Glomerular IgA deposition is a primary feature of immunoglobulin A vasculitis, and it may be indistinguishable from IgA nephropathy based on biopsy specimens, leading to speculation that immunoglobulin A vasculitis may be a systemic form of IgA nephropathy. However, immunoglobulin A vasculitis is clinically distinct from IgA nephropathy, usually manifesting as hematuria, purpuric rash, arthralgias, and abdominal pain.
Other serum immunologic tests are usually unnecessary. Complement concentrations are usually normal. Plasma IgA concentration may be elevated, and circulating IgA-fibronectin complexes are present; however, these findings are not helpful diagnostically.
Treatment of IgA Nephropathy
hypertension, serum creatinine > 1.2 mg/dL (106.08 micromol/L), or macroalbuminuria (urinary protein > 300 mg/day) and with a target urinary protein of < 500 mg/day
A sodium-glucose cotransporter 2 (SGLT2) inhibitor may be added for persistent proteinuria despite angiotensin inhibition
Corticosteroids for progressive disease, including increasing proteinuria, especially into the nephrotic range, and increasing serum creatinine level
Transplantation in advanced disease
Normotensive patients with intact renal function (serum creatinine < 1.2 mg/dL [106.08 micromol/L]) and only mild proteinuria (< 0.5 g/day) usually are not treated beyond angiotensin inhibition (with an ACE inhibitor or ARB) and an SGLT2 inhibitor. Patients with renal insufficiency or more severe proteinuria and hematuria are usually offered corticosteroids, which ideally should be started before significant renal insufficiency develops.
Angiotensin inhibition in IgA nephropathy
ACE inhibitors or ARBs are used on the premise that they reduce blood pressure, proteinuria, and glomerular fibrosis. Patients with the DD genotype for the ACE gene may be at greater risk of disease progression but may also be more likely to respond to ACE inhibitors or ARBs. For patients with hypertension, ACE inhibitors or ARBs are the antihypertensives of choice even for relatively mild chronic kidney disease.
Sodium-glucose cotransporter 2 inhibitors
An SGLT2 inhibitor may also be used for patients with proteinuria secondary to IgA nephropathy that has not improved with ACE inhibitors or ARBs. In a pre-specified subgroup analysis of patients with IgA nephropathy, SGLT2 inhibitors (which improve outcomes in patients with proteinuria due to diabetic kidney disease) reduced the risk of chronic kidney disease progression (1).
Corticosteroids and immunosuppressants in IgA nephropathy
For patients at high risk of disease progression (ie, proteinuria ≥ 1 g/day, an estimated glomerular filtration rate [eGFR] of 20 to 120 mL/min per 1.73 m2 after at least 3 months of supportive therapy), corticosteroids have been shown to slow the rate of progression to kidney failure (2). Higher doses corticosteroid regimens are associated with more serious adverse events (eg, infection requiring hospitalization).
Because of the risk of adverse effects, corticosteroids should probably be reserved for patients with any of the following:
Worsening or persistent proteinuria (> 1 g/day), especially if in the nephrotic range despite maximal ACE inhibitor or ARB therapy
Increasing serum creatinine level
Other treatments
For patients who progress to end-stage kidney disease, kidney transplantation is preferred over dialysis because of improved long-term disease-free survival. The condition recurs in approximately 30% of graft recipients (3).
Treatment references
1. Wheeler DC, Toto RD, Stefánsson BV, et alKidney Int 100(1):215-224, 2021. doi: 10.1016/j.kint.2021.03.033
2. Lv J, Wong MG, Hladunewich MA, et al: Effect of oral methylprednisolone on decline in kidney function or kidney failure in patients with IgA nephropathy: The TESTING randomized clinical trial. JAMA 327(19):1888-1898, 2022. doi: 10.1001/jama.2022.5368
3. Jäger C, Stampf S, Molyneux K, et al: Recurrence of IgA nephropathy after kidney transplantation: Experience from the Swiss transplant cohort study. BMC Nephrol 23(1):178, 2022. doi: 10.1186/s12882-022-02802-x
Prognosis for IgA Nephropathy
IgA nephropathy usually progresses slowly; renal insufficiency and hypertension develop within 10 years in 15 to 20% of patients. Progression to end-stage kidney disease occurs in 25% of patients after 20 years. When IgA nephropathy is diagnosed in childhood, prognosis is usually good. However, persistent hematuria invariably leads to hypertension, proteinuria, and renal insufficiency. Risk factors for progressive deterioration in renal function include the following:
Proteinuria > 1 g/day
Elevated serum creatinine level
Uncontrolled hypertension
Persistent microscopic hematuria
Extensive fibrotic changes in the glomerulus or interstitium
Crescents on biopsy
Key Points
IgA nephropathy is the most common cause of glomerulonephritis worldwide and is common among young adults and White and Asian people.
Consider the diagnosis in patients with unexplained signs of glomerulonephritis, particularly when it occurs within 2 days of a febrile mucosal illness or with flank pain.
Treat patients who have creatinine > 1.2 mg/dL (106.08 micromol/L) or proteinuria > 300 mg/day with ACE inhibitors or ARBs, and subsequently SGLT2 inhibitors if proteinuria persists.
Reserve corticosteroids for patients with worsening renal function or proteinuria (> 1 g/day) despite treatment with an ACE inhibitor or ARB plus an SGLT2 inhibitor.






