


Non-Hodgkin lymphomas are a heterogeneous group of disorders involving malignant, monoclonal proliferation of lymphoid cells in lymphoreticular sites, including lymph nodes, bone marrow, the spleen, the liver, and the gastrointestinal tract. Presenting symptoms usually include peripheral lymphadenopathy. However, some patients present without lymphadenopathy but with abnormal lymphocytes in circulation. Disease is likely to be disseminated at the time of presentation, and diagnosis is usually based on lymph node or bone marrow biopsy or both. Management strategies may include watch and wait, chemotherapy, targeted drugs (eg, kinase inhibitors), and immunotherapies (eg, monoclonal antibodies, chimeric antigen receptor T cells); occasionally, radiation therapy is added. With few exceptions, stem cell transplantation is usually reserved for patients with aggressive lymphomas after incomplete remission or relapse.
(See also Overview of Lymphomas.)
Non-Hodgkin lymphoma is more common than Hodgkin lymphoma. It is the sixth most common cancer in the United States and represents 4% of all new cancers in the United States each year and 3% of all cancer deaths. Over 80,000 new cases are diagnosed annually in all age groups, and there are about 20,000 deaths (1). Non-Hodgkin lymphoma is not one disease but rather a category of lymphocyte cancers with a number of subgroups largely divided into aggressive and indolent types. Incidence increases with age.
General reference
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin 2023;73(1):17-48. doi:10.3322/caac.21763
Etiology of Non-Hodgkin Lymphomas
The cause of non-Hodgkin lymphoma is unknown, although, as with the leukemias, substantial evidence suggests a viral cause (eg, human T-cell leukemia-lymphoma virus, Epstein-Barr virus, hepatitis B virus, hepatitis C virus, HIV, human herpesvirus 8) in some cases. Helicobacter pylori infection also increases lymphoma risk.
Patients at increased risk of non-Hodgkin lymphoma include those with
Secondary immunodeficiency (eg, when induced by immunosuppressants, such as those used in systemic rheumatic diseases and after solid organ transplant)
Autoimmune conditions (eg, rheumatoid arthritis, Sjogren syndrome)
Chronic inflammation and reactive lymph node hyperplasia
Possibly exposure to certain chemicals (eg, some herbicides and insecticides)
Non-Hodgkin lymphoma is one of the most common cancer in patients with HIV infection, and some patients with HIV present with lymphoma. Patients with non-Hodgkin lymphoma should generally be screened for HIV and hepatitis viruses.
Genetic factors appear to play a role. Certain single nucleotide polymorphisms increase the risk of lymphoma. Patients with a first-degree relative with Hodgkin or non-Hodgkin lymphoma have an increased risk of non-Hodgkin lymphoma.
Pathophysiology of Non-Hodgkin Lymphomas
Most non-Hodgkin lymphomas arise from B lymphocytes; the remainder arise from T lymphocytes or natural killer cells. The stage of lymphocyte differentiation at which the oncogenic event occurs determines the disease presentation and outcome.
Most lymphomas are nodal with variable involvement of the bone marrow and peripheral blood, although some lymphomas arise in or involve extranodal sites (eg, skin, gastrointestinal tract, lung, central nervous system). A leukemia-like picture with peripheral lymphocytosis and bone marrow involvement may be present in up to 50% of children and about 20% of adults with some types of non-Hodgkin lymphoma.
Hypogammaglobulinemia caused by a progressive decrease in immunoglobulin production is present in 15% of patients at diagnosis. Hypogammaglobulinemia increases the risk of serious bacterial infection, and patients may require IV immune globulin to replace deficient immunoglobulins.
Pearls & Pitfalls
|
Classification of Non-Hodgkin Lymphomas
Pathologic classification of non-Hodgkin lymphoma continues to evolve, reflecting new insights into the cells of origin and the biologic bases of these heterogeneous diseases. Since 2022, 2 classification systems are in existence: The 2022 WHO classification (1) and the 2022 International Consensus Classification (2). Both systems incorporate elements of morphology, immunophenotype, genetic information, and clinical patterns to subdivide and classify diseases into distinct entities with clinical relevance.
Non-Hodgkin lymphomas are commonly also categorized as indolent or aggressive:
Indolent: Slowly progressive and responsive to therapy but not typically curable with standard approaches
Aggressive: Rapidly progressive but responsive to chemotherapy and often curable
In children, non-Hodgkin lymphoma is almost always aggressive. Follicular and other indolent lymphomas are unusual. The treatment of these aggressive lymphomas (Burkitt, diffuse large B cell, and lymphoblastic lymphoma) presents special concerns, including gastrointestinal tract involvement (particularly in the terminal ileum); meningeal spread (requiring cerebrospinal fluid prophylaxis or treatment); and other sanctuary sites of involvement (eg, testes, brain). In addition, with these potentially curable lymphomas, treatment of adverse effects as well as outcome must be considered, including late risks of secondary cancer, cardiorespiratory sequelae, fertility preservation, and developmental consequences.
Classification references
1. Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms [published correction appears in Leukemia 2023 Sep;37(9):1944-1951]. Leukemia 2022;36(7):1720-1748. doi:10.1038/s41375-022-01620-2
2. Campo E, Jaffe ES, Cook JR, et al. The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee [published correction appears in Blood 2023 Jan 26;141(4):437]. Blood 2022;140(11):1229-1253. doi:10.1182/blood.2022015851
Symptoms and Signs of Non-Hodgkin Lymphomas
Most patients present with
Asymptomatic peripheral lymphadenopathy
Enlarged lymph nodes can be rubbery and discrete and later coalesce into masses. Affected nodes are usually not painful, unlike the tender nodes that often occur with viral infections. Nodal involvement is localized in some patients, but most patients have several areas affected. The initial physical examination should carefully look for nodes in the cervical, axillary, inguinal, and femoral regions.
In some patients, enlarged mediastinal and retroperitoneal nodes press on nearby structures, leading to symptoms. The most important of these are
Compression of the superior vena cava (SVC): Shortness of breath and facial edema (SVC syndrome)
Compression of the external biliary tree: Jaundice
Compression of the ureters: Hydronephrosis
Bowel obstruction: Vomiting and obstipation
Interference with lymph drainage: Chylous pleural or peritoneal fluid or lymphedema of a lower extremity
The skin is involved in some non-Hodgkin lymphomas. B-cell non-Hodgkin lymphoma can affect the scalp (follicular non-Hodgkin lymphoma) or the legs (large cell non-Hodgkin lymphoma), typically causing slightly raised, erythematous nodules. In cutaneous T-cell non-Hodgkin lymphoma, skin lesions can be diffuse, nonpalpable erythema or discrete papules, plaques, or tumors. In patients with dark skin, erythema may be subtle.

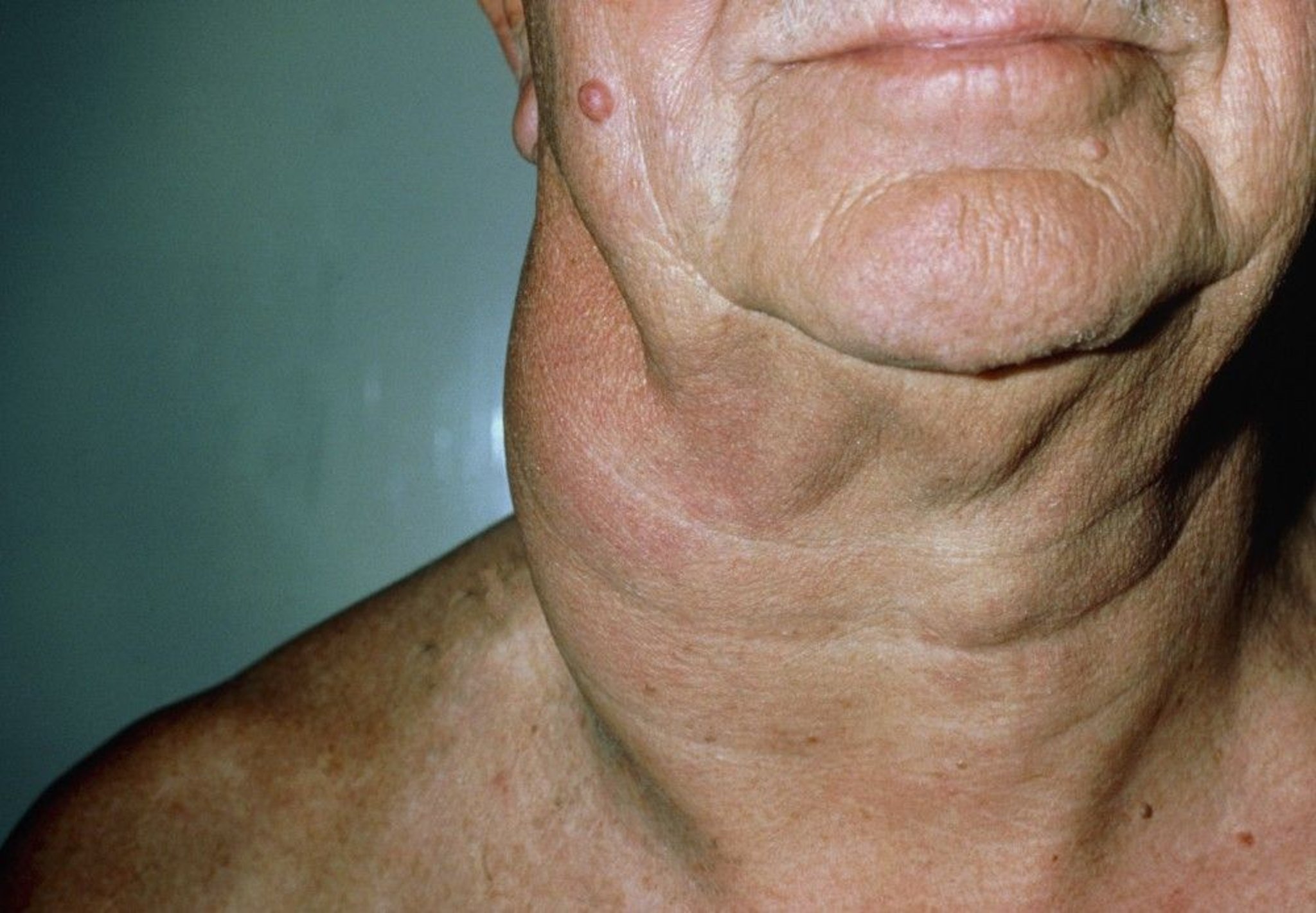
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
Systemic symptoms (eg, fatigue, fevers, night sweats, weight loss) can be the first manifestations in some patients, most commonly in aggressive lymphomas. These patients may not have noticed lymphadenopathy or not have external, palpable disease; these patients require CT or positron emission tomography (PET) imaging to discover the lesion(s).

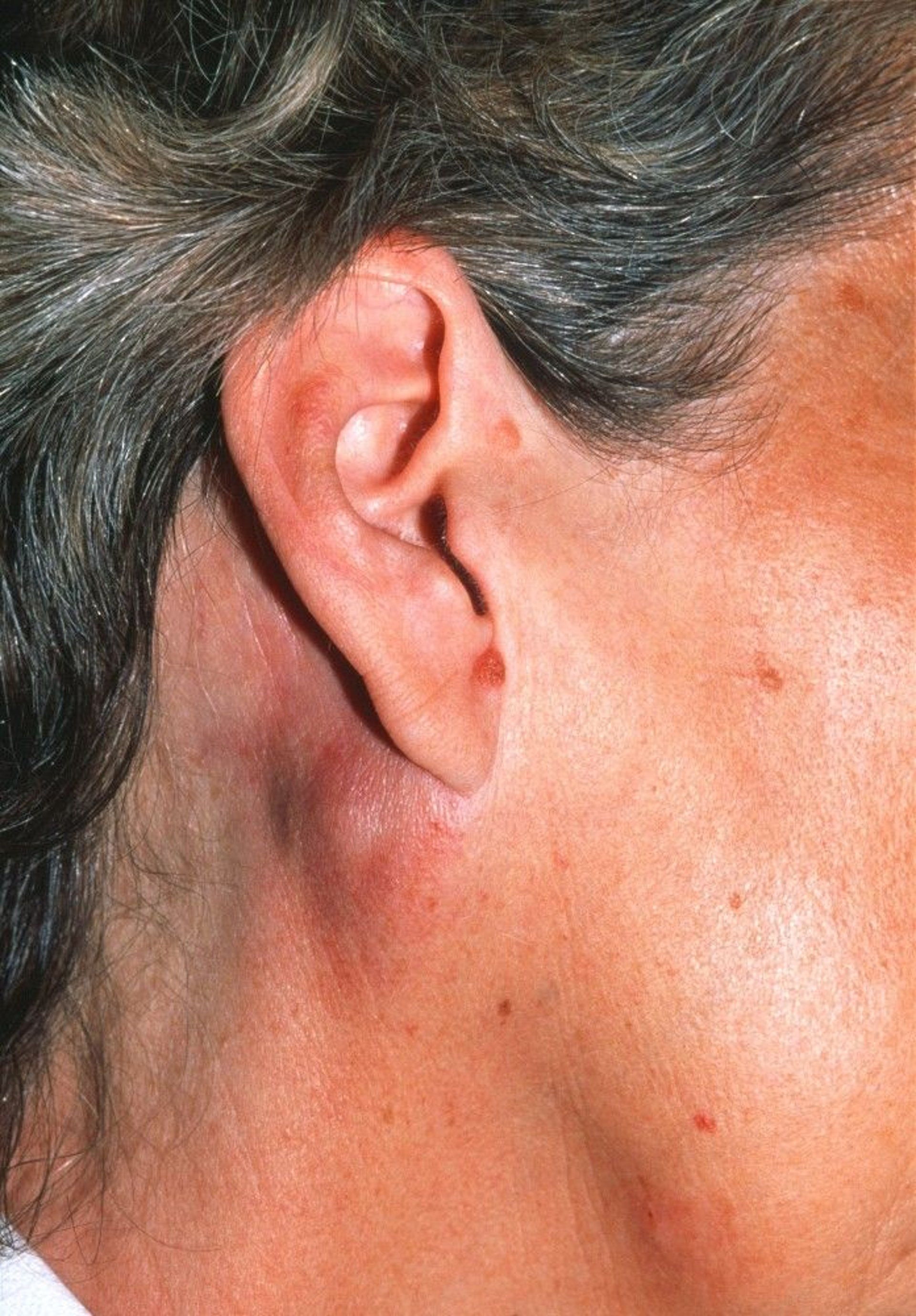
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
Anemia is initially present in some patients and eventually develops in many. It may be caused by
Bleeding due to gastrointestinal lymphoma, with or without low platelet levels
Hemolysis due to hypersplenism or Coombs’-positive hemolytic anemia
Bone marrow infiltration due to lymphoma
Bone marrow suppression due to chemotherapy or radiation therapy
Suppressed bone marrow function related to chronic inflammation
Manifestations of some specific lymphomas
Adult T-cell leukemia-lymphoma, which is associated with human T-lymphotropic virus 1 (HTLV-1), has a fulminating clinical course with skin infiltrates, lymphadenopathy, hepatosplenomegaly, and leukemia. The leukemic cells are malignant T cells, many with convoluted nuclei. Hypercalcemia often develops, related to humoral factors rather than to direct bone invasion.
Anaplastic large cell lymphoma may cause rapidly progressive skin lesions, adenopathy, and visceral lesions. This disease may be mistaken for Hodgkin lymphoma or metastatic undifferentiated carcinoma.
Diagnosis of Non-Hodgkin Lymphomas
Lymph node biopsy
Often unilateral bone marrow aspiration and biopsy
FDG-PET/CT of chest, abdomen, and pelvis for staging
MRI of brain and/or spinal cord if neurologic symptoms are present
As with Hodgkin lymphoma, non-Hodgkin lymphoma is usually suspected in patients with
Painless lymphadenopathy
Adenopathy detected on a chest radiograph or CT done for other reasons
Painless lymphadenopathy can also result from infectious mononucleosis, toxoplasmosis, cytomegalovirus infection, primary HIV infection, or acute leukemia.
Similar chest radiograph findings can result from lung carcinoma, sarcoidosis, or tuberculosis.
Less commonly, patients present after a finding of peripheral lymphocytosis on a complete blood count (CBC) done for nonspecific symptoms. In such cases, the differential diagnosis includes leukemia, Epstein-Barr virus infection, and Duncan syndrome (X-linked lymphoproliferative syndrome).
Tests needed to make the diagnosis are followed by tests to complete staging and assess etiology and prognosis (1).
Diagnostic tests
Enlarged lymph nodes are biopsied. If a node is palpable, no imaging is required initially, although CT or ultrasonography may be needed to properly plan subsequent tests.
If the lesion is easily palpable, an open biopsy is preferred. If the lesion is in the lung or abdomen, a core needle biopsy (18- to 20-gauge needle) done using CT or ultrasound guidance can often obtain an adequate specimen for diagnosis. A fine needle biopsy (percutaneous or bronchoscopic) frequently will not produce adequate tissue, especially for initial diagnosis; core biopsy is preferred if deemed safe.
Biopsy samples should be reviewed by a pathologist with expertise in lymphoma diagnosis so that the lymphoma can be correctly classified. If this review is not available locally, the slides should be sent to a reference laboratory with hematopathology expertise. The proper classification of non-Hodgkin lymphoma is critical for treatment planning. Non-Hodgkin lymphomas are potentially curable, but without a precise diagnosis, optimal therapy may not be chosen.
Histologic criteria on biopsy include destruction of normal lymph node architecture and invasion of the capsule and adjacent fat by characteristic neoplastic cells.
Immunophenotyping studies (using immunohistochemistry or flow cytometry) to determine the cell of origin are of great value in identifying specific subtypes and helping define prognosis and management; these studies also can be done on peripheral cells if they are present, but typically these stains are applied to formalin-fixed, paraffin-embedded tissue.
Demonstration of the leukocyte common antigen CD45 by immunoperoxidase rules out metastatic cancer, which is often in the differential diagnosis of “undifferentiated” cancers. The test for leukocyte common antigen, most surface marker studies, and gene rearrangement (to document B-cell or T-cell clonality) can be done on fixed tissues. Cytogenetics and flow cytometry require fresh tissue.
Next generation sequencing may hold diagnostic or prognostic significance in cases of non-Hodgkin lymphoma and can be performed on fresh or fixed tissues (assay dependent).
Staging tests
Once the diagnosis of lymphoma is made, staging tests are done.
A combined fluorodeoxyglucose (FDG)-PET/CT scan of the chest, abdomen, and pelvis is recommended. PET/CT provides accurate location of lesions, their size (from CT) and tumor metabolism (from FDG-PET). If combined FDG-PET/CT is not available, a contrast-enhanced CT scan of the chest, abdomen, and pelvis is done.
Unilateral bone marrow aspiration and biopsy is often done in patients with non-Hodgkin lymphoma. While marrow evaluation may be of diagnostic value, its utility in staging and prognosis in most lymphomas is less clear. Bone marrow assessment may be of limited value in settings where marrow involvement is unlikely (eg, early-stage diffuse large B-cell lymphoma) or in settings where results would not likely influence management (eg, advanced-stage disease).
Testing for complications and prognosis
Blood tests typically include complete blood count with white blood cell differential, kidney function and liver tests (including serum creatinine, bilirubin, calcium, aspartate aminotransferase, albumin, alkaline phosphatase, and lactate dehydrogenase), uric acid, beta-2 microglobulin, and vitamin D levels. Serum protein electrophoresis with IgG, IgA, and IgM immunoglobulin levels are also done.
Other tests are done depending on findings (eg, MRI of brain and/or spinal cord for neurologic symptoms). If uric acid levels are high, serum glucose-6-phosphate dehydrogenase (G6PD) level is checked because G6PD deficiencytumor lysis syndrome but may cause hemolytic anemia in patients with G6PD deficiency.
Testing for etiology
Patients with non-Hodgkin lymphoma are initially screened for HIV and hepatitis B and C viruses. Patients diagnosed with adult T-cell leukemia/lymphoma (ATLL) are also checked for human T-cell lymphotropic virus type 1 (HTLV-1).
Staging
After diagnosis, stage is determined to guide therapy. The commonly used Lugano staging system (see table Lugano Staging of Hodgkin Lymphoma and Non-Hodgkin Lymphoma) incorporates
Symptoms
Physical examination findings
Results of imaging tests, including CT of the chest, abdomen, and pelvis, and functional imaging with FDG-PET
Bone marrow biopsy (in selected cases)
Although stage I non-Hodgkin lymphoma does occur, the disease is typically disseminated when first recognized.
Diagnosis reference
1. Cheson BD, Fisher RI, Barrington SF, et al: Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: The Lugano classification. J Clin Oncol 32(27):3059–3068, 2014.
Treatment of Non-Hodgkin Lymphomas
Watch and wait (for indolent, largely asymptomatic lymphomas)
Chemotherapy
Radiation therapy (most common in patients with limited-stage disease and sometimes in those with advanced-stage disease)
Immunotherapy (eg, monoclonal antibodies or antibody-drug conjugates targeting CD20, CD19, or CD79; bispecific antibodies targeting CD20 and CD3; or chimeric antigen receptor T cells [CAR T cells])
Targeted drugs (eg, BTK [Bruton tyrosine kinase] inhibitors, PI3K [phosphoinositide 3-kinase] inhibitors, cereblon inhibitors, EZH2 [enhancer of zeste homolog 2] inhibitors, XPO1 [exportin 1] inhibitors)
Sometimes hematopoietic stem cell transplantation (autologous or allogeneic)
Treatment varies considerably with cell type, which are too numerous to permit detailed discussion. Generalizations can be made regarding limited versus advanced disease and aggressive versus indolent forms. Burkitt lymphoma and cutaneous T-cell lymphomas are discussed separately. For patients with indolent lymphomas and no significant signs or symptoms of lymphoma, a "watch and wait" approach (withholding treatment while closely monitoring) can be used.
Limited disease (stages I-II)
For stage I indolent non-Hodgkin lymphoma (uncommon because most patients have stage II to IV when diagnosed), external beam radiation therapy can be the sole initial treatment. Regional radiation therapy may offer long-term control and possibly cure in about 40% of patients with stage I disease (1, 2). Stage II indolent non-Hodgkin lymphoma is most commonly treated as advanced-stage disease.
Limited-stage aggressive non-Hodgkin lymphomas can be managed with a combination of chemotherapy plus radiation therapy or with chemotherapy alone (plus anti-CD20 monoclonal antibodies for B-cell lymphomas).
Advanced disease (stages II-IV)
Stage II non-Hodgkin lymphoma is managed as advanced stage disease in many circumstances. Most patients with all types of non-Hodgkin lymphoma who have stage II to IV disease are candidates for chemoimmunotherapy. In these cases, radiation therapy may be used to limit the number of cycles of chemoimmunotherapy or provide localized treatment for residual sites of bulk disease.
For indolent lymphomas,
In patients with aggressive B-cell lymphomas3). These results vary significantly by International Prognostic Index (IPI) for diffuse large B-cell lymphoma score. Patients who are disease free at ≥3).
The approach in peripheral T-cell non-Hodgkin lymphoma and primary central nervous system lymphoma is different. In these patients, autologous stem cell transplantation may be offered to initial responders before relapse occurs with the intention of improving the likelihood of cure. In autologous stem cell transplantation, stem cells are obtained from the patient by peripheral blood leukopheresis and are transfused back into the patient after high-dose chemotherapy. Similarly, in some younger patients with mantle cell lymphoma who have responded to initial therapy, autologous stem cell transplantation may be done to prolong remission.
Lymphoma relapse
Patients with aggressive non-Hodgkin lymphoma who are not in remission at end of therapy or who relapse are treated with second-line chemotherapy regimens followed by autologous stem cell transplantation if they are relatively young and in good health. In some patients at very high risk of relapse as well as in those for whom autologous transplant is not feasible or has already failed, stem cells from a matched sibling or unrelated donor (allogeneic transplants) can be effective. In general, the older the patient, the less likely an allogeneic transplantation will be offered because older patients have higher rates of transplantation complications.
Patients with diffuse large B-cell lymphoma (DLBCL) who have persistent lymphoma or progression within 12 months of completion of frontline immunochemotherapy as well as those with persistent lymphoma despite at least 2 prior lines of therapy may be candidates for chimeric antigen receptor (CAR) T cells. CAR T cells are T cells (most commonly autologous T cells) that have been genetically engineered to recognize a tumor antigen (eg, CD19). After infusion, they undergo activation and expansion. Approximately 60% of patients who receive CAR T cells as second-line therapy and approximately 30% of patients undergoing CAR T therapy as a subsequent treatment achieve a durable response from this therapy (4, 5, 6, 7, 8).
Patients not eligible for either stem cell transplantation or CAR T cells, or for whom these treatments have failed, may receive treatment with various therapies, mostly for palliation. These therapies vary widely and are constantly changing as new treatments are developed.
In indolent lymphomas, patients may be managed using a wide variety of strategies depending on
Lymphoma-related factors (eg, histopathology, stage, molecular characteristics, immunologic characteristics)
Patient-related factors (eg, age, comorbidities)
The type of and response to prior therapy.
Many of the same agents used for first-line treatment may be given to patients in relapse. In some cases, the same treatment may be repeated if it was previously effective and well tolerated. High-dose chemotherapy combined with autologous stem cell transplantation is used occasionally in patients who have high-risk lymphoma biology (including a poor response to chemotherapy), and although cure remains unlikely, remission may be superior to that with secondary palliative therapy alone. Reduced intensity allogeneic transplantation is a potentially curative option in some patients with indolent lymphoma. The mortality rate of patients undergoing myeloablative transplantation has decreased dramatically.
Complications of treatment
An immediate complication of most therapies is infection that occurs during periods of neutropenia. Although use of growth factors that stimulate white blood cell production has helped, infection continues to pose a problem.
The gastrointestinal adverse effects of chemotherapy can be largely relieved or prevented by antiemetics and bowel programs.
Patients receiving anthracyclines are at risk of cardiomyopathy and/or arrhythmias.
After successful treatment, patients should be referred to a cancer survivorship clinic for a care plan that can be implemented by the patient's primary care team. This plan is tailored to the patient's comorbidities and risks specific to the treatment they received.
Chemotherapy and radiation therapy have late complications. In the first 10 years after treatment, there is a risk of myelodysplasia or acute leukemia due to bone marrow damage from certain chemotherapy agents. After 10 years, the risk of secondary cancers increases, especially in patients who received radiation to the chest.
Treatment references
1. Lo AC, Campbell BA, Pickles T, et al. Long-term outcomes for patients with limited-stage follicular lymphoma: update of a population-based study. Blood 2020;136(8):1006-1010. doi:10.1182/blood.2019004588
2. Wilder RB, Jones D, Tucker SL, et al. Long-term results with radiotherapy for Stage I-II follicular lymphomas. Int J Radiat Oncol Biol Phys 2001;51(5):1219-1227. doi:10.1016/s0360-3016(01)01747-3
3. Tilly H, Morschhauser F, Sehn LH, et al. Polatuzumab Vedotin in Previously Untreated Diffuse Large B-Cell Lymphoma. N Engl J Med 2022;386(4):351-363. doi:10.1056/NEJMoa2115304
4. Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 2020;396(10254):839-852. doi:10.1016/S0140-6736(20)31366-0
5. Abramson JS, Solomon SR, Arnason J, et al. Lisocabtagene maraleucel as second-line therapy for large B-cell lymphoma: primary analysis of the phase 3 TRANSFORM study. Blood 2023;141(14):1675-1684. doi:10.1182/blood.2022018730
5. Locke FL, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N Engl J Med 2022;386(7):640-654. doi:10.1056/NEJMoa2116133
7. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 2017;377(26):2531-2544. doi:10.1056/NEJMoa1707447
8. Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med 2019;380(1):45-56. doi:10.1056/NEJMoa1804980
Prognosis for Non-Hodgkin Lymphomas
Prognosis varies by the type and stage of lymphoma and individual patient factors. In general, patients with peripheral T-cell or natural killer (NK)/T-cell lymphomas typically have a worse prognosis than those with B-cell non-Hodgkin lymphoma. Within each non-Hodgkin lymphoma variant, prognosis is related to differences in tumor cell biology.
The most commonly used prognostic scoring system is the International Prognostic Index (IPI) for diffuse large B-cell lymphoma. However, the IPI score is used only for diffuse large B-cell lymphoma (DLBCL). There are also scoring systems for follicular lymphoma (FLIPI) and mantle cell lymphoma (MIPI). Online calculators are available to estimate prognosis in other types of non-Hodgkin lymphoma as well.
The IPI considers 5 risk factors:
Age > 60 years
Poor performance status (can be measured using the Eastern Cooperative Oncology Group tool)
Elevated lactate dehydrogenase (LDH) level
> 1 extranodal site
Stage III or IV disease
Outcome is worse with an increasing number of risk factors. Patients without any of the risk factors have a very high cure rate. The original IPI score uses the 5 factors as discrete variables (eg, either age over 60 years or under 60 years). A modification, the Diffuse Large B-cell Lymphoma Prognosis (IPI24), which calculates the chance of being disease free at 24 months from diagnosis, includes the above factors as continuous variables and also includes absolute lymphocyte count.
Key Points
Non-Hodgkin lymphomas are a group of related cancers involving lymphocytes; they vary significantly in their rate of growth and response to treatment.
The disease is usually already disseminated at the time of diagnosis.
Molecular and genetic tests are essential for diagnosis and management.
Limited indolent disease may be treated with radiation therapy.
Treat more advanced disease (indolent or aggressive) with immunotherapy, chemotherapy, hematopoietic stem cell transplantation, or a combination depending on the type and stage of non-Hodgkin lymphoma.
More Information
The following English language resource provides information for clinicians and support and information for patients. THE MANUAL is not responsible for the content of this resource.
Leukemia & Lymphoma Society: Resources for Healthcare Professionals: provides educational resources for health care practitioners as well as information for patient referrals






