


Ataxia-telangiectasia results from a DNA repair defect that frequently results in humoral and cellular immunodeficiency; it causes progressive cerebellar ataxia, oculocutaneous telangiectasias, and recurrent sinopulmonary infections. IgA and serum alpha-1 fetoprotein levels are measured. Genetic testing is needed to confirm the diagnosis. Treatment is with prophylactic antibiotics or with immune globulin.
(See also Overview of Immunodeficiency Disorders and Approach to the Patient With an Immunodeficiency Disorder.)
Ataxia-telangiectasia is an autosomal-recessive primary immunodeficiency disorder that involves combined humoral and cellular deficiencies. Estimated incidence is 1 in 20,000 to 100,000 births. Ataxia-telangiectasia is caused by mutations in the gene that encodes ataxia-telangiectasia–mutated (ATM) protein. ATM is involved in detection of DNA damage and helps control the rate of cell growth and division.
Patients often lack IgA and IgE and have a progressive T-cell defect.
Symptoms and Signs of Ataxia-Telangiectasia
Age at onset of neurologic symptoms and evidence of immunodeficiency vary.
Ataxia is frequently the first symptom and usually develops when children begin to walk. Progression of neurologic symptoms leads to severe disability. Speech becomes slurred, choreoathetoid movements and nystagmus develop, and muscle weakness usually progresses to muscle atrophy.
Telangiectasias may not appear until age 4 to 6 years; they are most prominent on the bulbar conjunctivae, ears, antecubital and popliteal fossae, and sides of the neck.
Recurrent sinopulmonary infections lead to recurrent pneumonia, bronchiectasis, and chronic restrictive pulmonary disease.
Certain endocrine abnormalities (eg, gonadal dysgenesis, testicular atrophy, diabetes mellitus) may occur.
Frequency of cancer (especially leukemia, lymphoma, brain tumors, and gastric cancer) is high. Cancer typically occurs after age 10 and at a rate of about 1%/year but is a lifelong risk and can occur at any age.

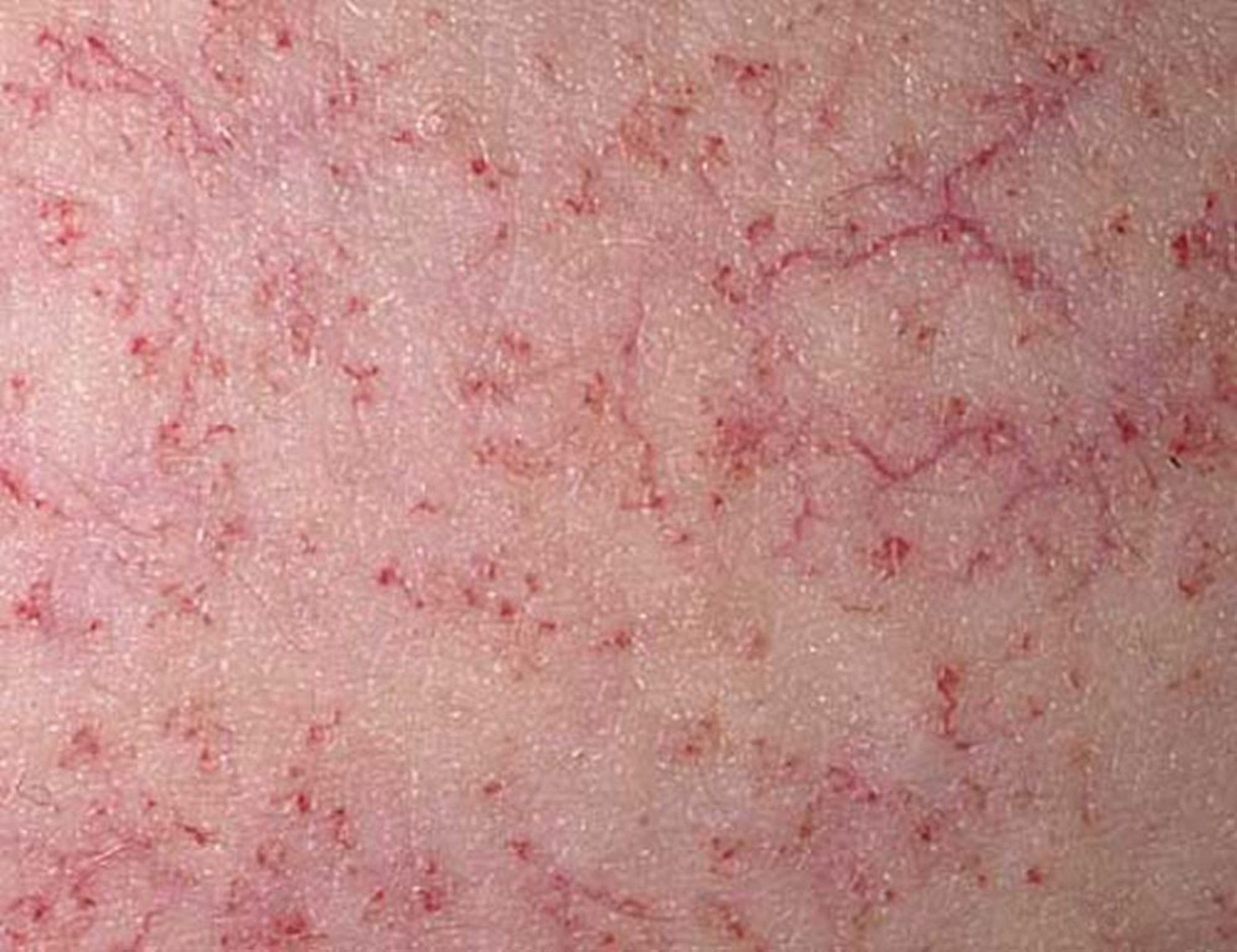
Photo provided by Thomas Habif, MD.
Diagnosis of Ataxia-Telangiectasia
IgA and serum alpha-1 fetoprotein levels
Genetic testing
The following clinical findings suggest the diagnosis of ataxia-telangiectasia:
Cerebellar ataxia (particularly when telangiectasias are present)
Low levels of IgA (present in 80% of patients with this disorder)
High levels of serum alpha-1 fetoprotein
If karyotype analysis is done, chromosome breaks, consistent with a defect in DNA repair, are often identified.
Diagnosis of ataxia-telangiectasia is confirmed by identifying mutations on both alleles of the gene for ATM protein. Because carriers of an ataxia-telangiectasia mutation usually remain asymptomatic, testing siblings for a carrier state can help predict their chance of having an affected child.
Testing for endocrine abnormalities and cancers is done based on clinical presentation.
Family members of known patients should be offered genetic counseling with the option to test for the ataxia-telangiectasia carrier state because carriers may be at increased risk for cancer (1))
Diagnosis reference
1. Rothblum-Oviatt C, Wright J, Lefton-Greif MA, et al: Ataxia telangiectasia: a review. Orphanet J Rare Dis 11(1):159, 2016. doi:10.1186/s13023-016-0543-7
Treatment of Ataxia-Telangiectasia
Supportive care using prophylactic antibiotics or immune globulin (IgG) replacement therapy
Treatment with prophylactic antibiotics or immune globulin may help patients with ataxia-telangiectasia.
1).
Chemotherapy is often indicated for treatment of associated cancers.
Treatment reference
1. Nissenkorn A, Hassin-Baer S, Lerman SF, et alJ Child Neurol 28(2):155–160, 2013. doi:10.1177/0883073812441999
Key Points
Ataxia-telangiectasia involves combined humoral and cellular deficiencies.
Ataxia is frequently the first symptom and usually develops when children begin to walk.
There is no effective treatment for the progressive neurologic deterioration, which causes death, usually by age 30.






