


Marfan syndrome consists of connective tissue anomalies resulting in ocular, skeletal, and cardiovascular abnormalities (eg, dilation of ascending aorta, which can lead to aortic dissection). Diagnosis is clinical. Treatment may include prophylactic beta-blockers to slow dilation of the ascending aorta and prophylactic aortic surgery.
Inheritance of Marfan syndrome is autosomal dominant. The basic molecular defect results from mutations in the gene encoding the glycoprotein fibrillin-1 (FBN1), which is the main component of microfibrils and helps anchor cells to the extracellular matrix. The principal structural defect involves the cardiovascular, musculoskeletal, and ocular systems. The pulmonary system and central nervous system are also affected.
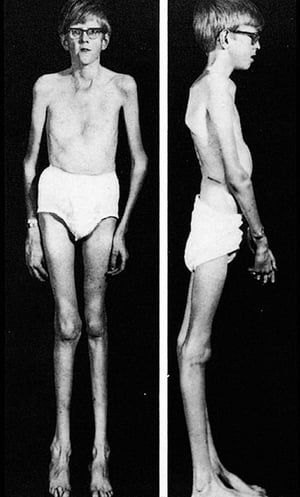
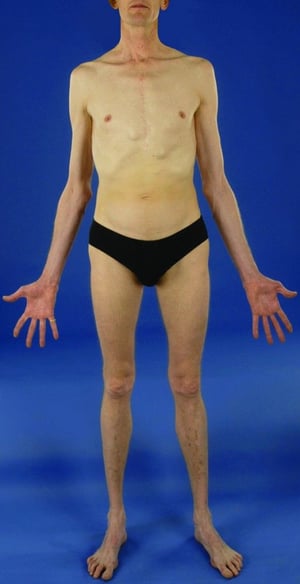
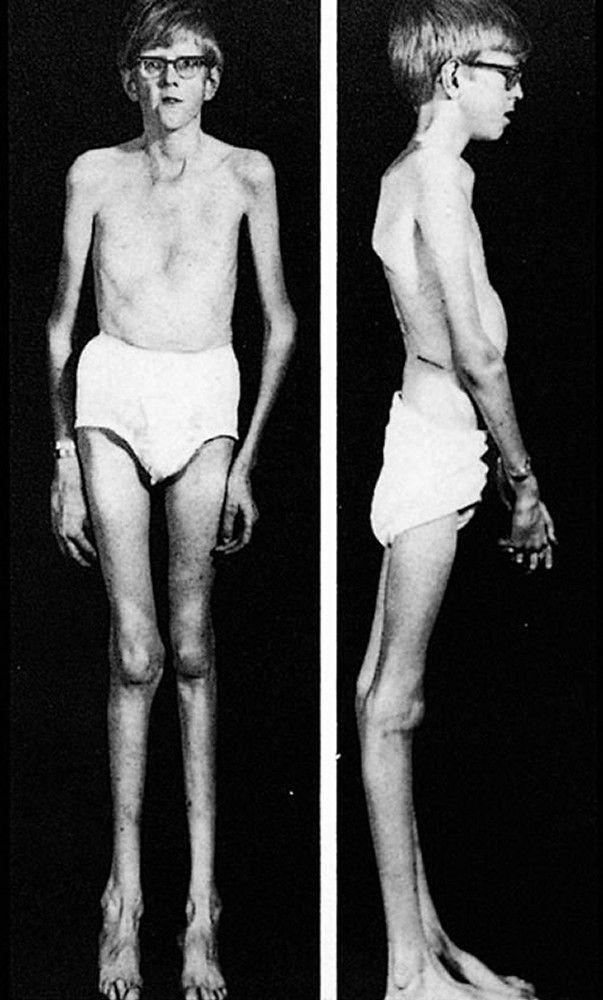
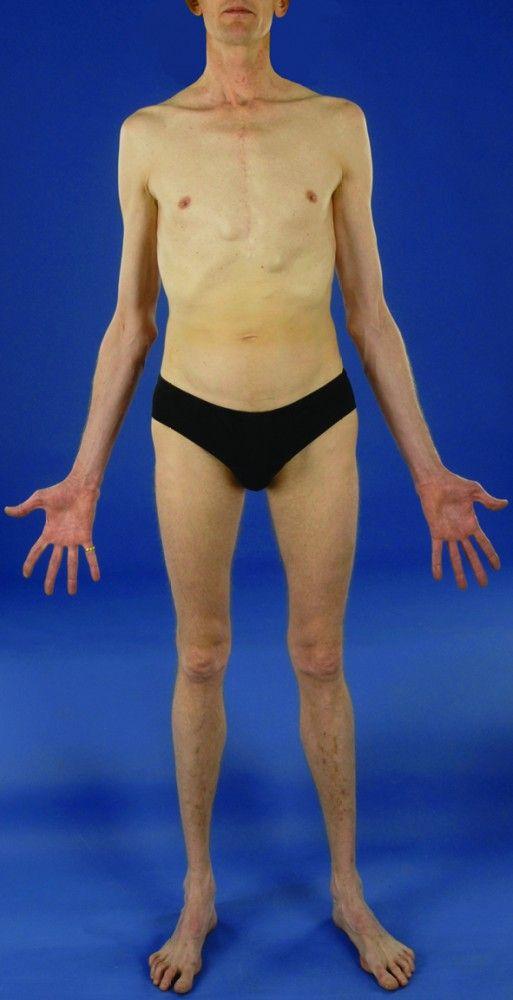
There are many different manifestations of the genetic mutation that causes Marfan syndrome; however, it is typically recognized by the constellation of long limbs, aortic root dilation, and dislocated lenses.
Symptoms and Signs of Marfan Syndrome
Cardiovascular system
Major findings include
Valvular prolapse
Most severe complications result from pathologic changes in the aortic root and ascending aorta. The aortic media is affected preferentially in areas subject to the greatest hemodynamic stress. The aorta progressively dilates or acutely dissects, beginning in the coronary sinuses, sometimes before 10 years of age. The aortic root dilates in 50% of children and in 60 to 80% of adults and can cause aortic regurgitation, in which case a diastolic murmur may be heard over the aortic valve.
Redundant cusps and chordae tendineae may lead to mitral valve prolapse or regurgitation; mitral valve prolapse may cause a systolic click and a late systolic murmur or, in severe cases, a holosystolic murmur.
Bacterial endocarditis may develop in affected valves.
Musculoskeletal system
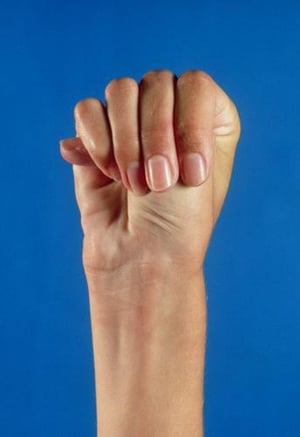
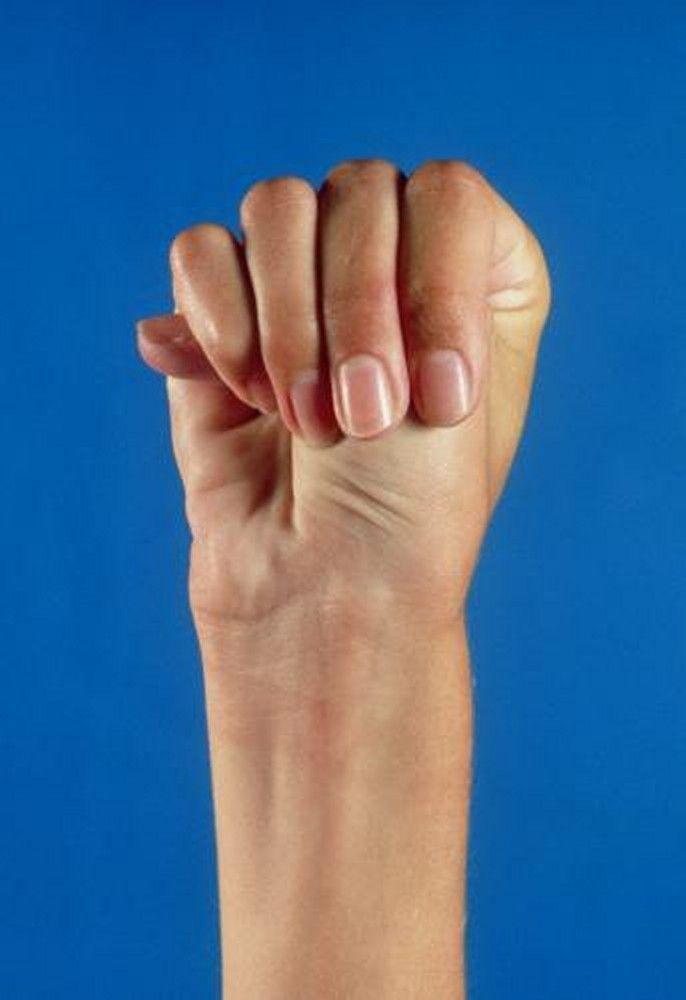
Severity varies greatly. Patients are taller than average for age and family; arm span exceeds height. Arachnodactyly (disproportionately long, thin digits) is noticeable, often by the thumb sign (the distal phalanx of the thumb protrudes beyond the edge of the clenched fist). Sternum deformity—pectus carinatum (outward displacement) or pectus excavatum (inward displacement)—is common, as are joint hyperextensibility (but usually small flexion contractures to the elbows), genu recurvatum (backward curvature of the legs at the knees), pes planus (flat feet), kyphoscoliosis, and diaphragmatic and inguinal hernias. Subcutaneous fat usually is sparse. The palate is often high-arched.
By permission of the publisher. From Macro R: Atlas of Heart Diseases: Congenital Heart Disease. Edited by E Braunwald (series editor) and RM Freedom. Philadelphia, Current Medicine, 1997.
© Springer Science+Business Media
MEDICAL PHOTO NHS LOTHIAN/SCIENCE PHOTO LIBRARY

Photo courtesy of David D. Sherry, MD.

By permission of the publisher. From Macro R: Atlas of Heart Diseases: Congenital Heart Disease. Edited by E Braunwald (series editor) and RM Freedom. Philadelphia, Current Medicine, 1997.

© Springer Science+Business Media

MEDICAL PHOTO NHS LOTHIAN/SCIENCE PHOTO LIBRARY

Photo courtesy of David D. Sherry, MD.
Ocular system
Findings include ectopia lentis (subluxation or upward dislocation of the lens) and iridodonesis (tremulousness of the iris). The margin of the dislocated lens can often be seen through the undilated pupil. High-grade myopia may be present, and spontaneous retinal detachment may occur.
Pulmonary system
Cystic lung disease and recurrent spontaneous pneumothorax may occur. These disorders can cause pain and shortness of breath.
Central nervous system
Dural ectasia (widening of the dural sac surrounding the spinal cord) is a common finding and most frequently occurs in the lumbosacral spine. It may cause headache, lower back pain, or neurologic deficits manifested by bowel or bladder weakness.
Diagnosis of Marfan Syndrome
Clinical criteria
Genetic testing
Echocardiography/MRI (measurement of the aortic root, detection of valve prolapse)
Slit-lamp examination (lens abnormalities)
X-rays of skeletal system (hand, spine, pelvis, chest, foot, and skull for characteristic abnormalities)
MRI of the lumbosacral spine (dural ectasia)
Diagnosis of Marfan syndrome can be difficult because many patients have only a few typical symptoms and signs and no specific histologic or biochemical changes. Considering this variability, diagnostic criteria are based on constellations of clinical findings and family and genetic history. (For more on diagnosis, see the revised Ghent nosology [2010], which heavily weights the presence of aortic root dilation or dissection, and lens dislocation.) Nonetheless, diagnosis is uncertain in many partial cases of Marfan syndrome.
Homocystinuria can partially mimic Marfan syndrome but can be differentiated by detecting homocystine in the urine. Genetic testing for FBN1 mutations can help establish the diagnosis in people who do not meet all clinical criteria, but FBN1 mutation-negative cases exist. Prenatal diagnosis by analysis of the FBN1 gene is hampered by poor genotype/phenotype correlation (> 1700 different mutations have been described).
Standard imaging of the skeletal, cardiovascular, and ocular systems is done to detect any clinically relevant structural abnormalities and to provide information contributing to the diagnostic criteria (eg, echocardiography to identify aortic root enlargement).
In addition to the criteria established within organ systems, family history (first-degree relative with Marfan syndrome) and genetic history (presence of the FBN1 mutation known to cause Marfan syndrome) are considered major criteria.
Prognosis for Marfan Syndrome
Advancements in therapy and regular monitoring have improved quality of life and reduced mortality. Median life expectancy increased from 48 years in 1972 to near normal today in people receiving appropriate medical care. However, life expectancy is still reduced for the average patient, primarily because of the cardiac and vascular complications. This decreased life expectancy can take an emotional toll on an adolescent and the family.
Treatment of Marfan Syndrome
Induction of precocious puberty in tall girls
Beta-blockers
Elective aortic repair and valve repair
Bracing and surgery for scoliosis
Treatment of Marfan syndrome is focused on prevention and treatment of complications.
Prophylactic surgery is offered if aortic diameter is > 5 cm (less in children). Pregnant women are at especially high risk of aortic complications; elective aortic repair before conception should be discussed. Severe valve regurgitation is also surgically repaired. Bacterial endocarditis prophylaxis before invasive procedures is not indicated except in patients who have prosthetic valves or who previously had infective endocarditis ( see Table: Procedures Requiring Antimicrobial Endocarditis Prophylaxis in High-Risk Patients in the US and see Table: Recommended Endocarditis Prophylaxis During Oral-Dental or Respiratory Tract Procedures*).
Scoliosis is managed with bracing as long as possible, but surgical intervention is encouraged in patients with curves of 40 to 50°.
Cardiovascular, skeletal, and ocular findings (including echocardiography) should be reevaluated annually.
Appropriate genetic counseling is indicated.
Key Points
Marfan syndrome results from an autosomal dominant mutation of the gene encoding the glycoprotein fibrillin-1, which is the main component of microfibrils, resulting in numerous possible deformities and defects.
Manifestations vary widely, but the principal structural defects involve the cardiovascular, musculoskeletal, and ocular systems, causing a typical constellation of long limbs, aortic root dilation, and dislocated lenses.
Aortic dissection is the most dangerous complication.
Diagnose using clinical criteria; genetic testing is often done.
Do imaging tests of the skeletal, cardiovascular, and ocular systems to detect structural abnormalities.
Give all patients a beta-blocker to help prevent aortic complications; treat other complications as they arise.
More Information
The following English-language resource may be useful. Please note that THE MANUAL is not responsible for the content of this resource.






