




En el síndrome de Bartter y en el síndrome de Gitelman, una anomalía hereditaria de los túbulos renales provoca que los riñones eliminen cantidades excesivas de electrólitos (potasio, sodio y cloruro), lo cual ocasiona anomalías del crecimiento, electrolíticas y en ocasiones neurológicas y musculares.
(Véase también Introducción a los trastornos congénitos de los túbulos renales.)
El síndrome de Bartter y el síndrome de Gitelman son hereditarios y suelen estar causados por un gen recesivo ( ver figura Trastornos autosómicos recesivos (o no ligados al cromosoma X)). Así pues, una persona con el síndrome de Bartter o el síndrome de Gitelman suele haber heredado dos genes recesivos alterados, uno de cada progenitor. Debido a que se necesitan dos genes cuando está involucrado un gen recesivo, los padres son portadores del gen pero no tienen el síndrome. Sin embargo, los hermanos de niños con el trastorno podrían tenerlo. Aunque ambos son raros, el síndrome de Gitelman es más común que el síndrome de Bartter.
En el síndrome de Bartter y en el síndrome de Gitelman, los riñones no pueden reabsorber sal (cloruro de sodio) normalmente desde el túbulo renal. Así pues, los riñones excretan cantidades excesivas de los electrólitos sodio y cloruro por la orina. La pérdida de sodio y cloruro conduce a una producción de orina excesiva y en consecuencia a una deshidratación crónica leve.
La deshidratación leve hace que el cuerpo produzca más cantidad de la enzima renina y de la hormona aldosterona, una hormona que ayuda a regular la presión arterial. El incremento de aldosterona aumenta la concentración de potasio y la secreción de ácido en los riñones, lo que conduce a un bajo nivel de potasio en sangre (hipocaliemia) y a una pérdida de ácidos en la sangre que provoca que el pH sanguíneo sea alcalino (un trastorno denominado alcalosis metabólica). Aunque los túbulos se ven afectados en ambos síndromes, los riñones por lo demás no están afectados y filtran los productos de desecho con normalidad.
Las vías urinarias

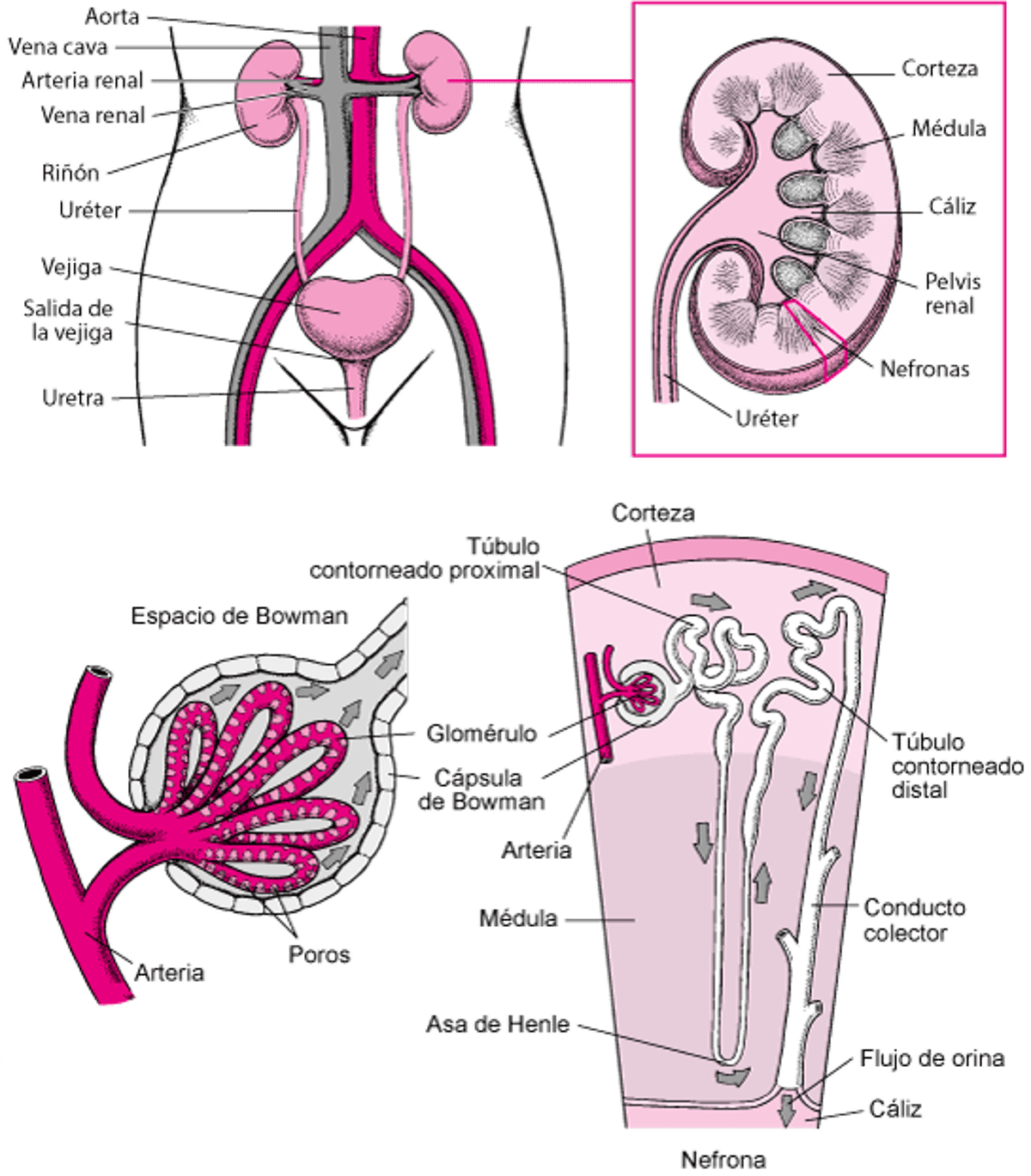
Las principales diferencias entre los dos síndromes son
Los genes que están involucrados
La parte del túbulo que se ve afectada
La edad a la que comienzan los síntomas
Síntomas del síndrome de Bartter y del síndrome de Gitelman
Los síntomas del síndrome de Bartter pueden aparecer antes del nacimiento, durante el periodo de lactancia o en los primeros años de la infancia.
Los síntomas del síndrome de Gitelman pueden aparecer en un periodo comprendido entre los últimos años de la niñez y la edad adulta.
Sin embargo, algunas personas, en particular las que tienen el síndrome de Gitelman, no presentan síntomas y se diagnostican solo después de realizar análisis de sangre por otras razones.
Los niños con estos síndromes tienen síntomas que son similares a los de las personas que toman diuréticos, unos fármacos que aumentan la producción de orina y pueden causar desequilibrios químicos en la sangre. Sin embargo, a diferencia de lo que ocurre con los diuréticos, en el síndrome de Bartter o en el síndrome de Gitelman, los síntomas pueden no cesar al solo interrumpir el fármaco.
Los fetos con síndrome de Bartter pueden presentar retraso del crecimiento en el útero. Algunos niños nacen de forma prematura y pueden tener discapacidad intelectual.
Los niños con este síndrome, y a veces los que padecen el síndrome de Gitelman, pueden tener un crecimiento deficiente y retraso en el desarrollo. La pérdida de magnesio, calcio o potasio puede ocasionar debilidad muscular, calambres, espasmos, o fatiga, especialmente en los sujetos con síndrome de Gitelman. Los niños con cualquiera de estos dos síndromes pueden tener sed excesiva, pueden producir grandes cantidades de orina, pueden desear sal y pueden tener náuseas y vómitos.
La pérdida de sodio y cloro conduce a una deshidratación crónica leve. Las personas con síndrome de Bartter o el síndrome de Gitelman pueden tener presión arterial baja (hipotensión).

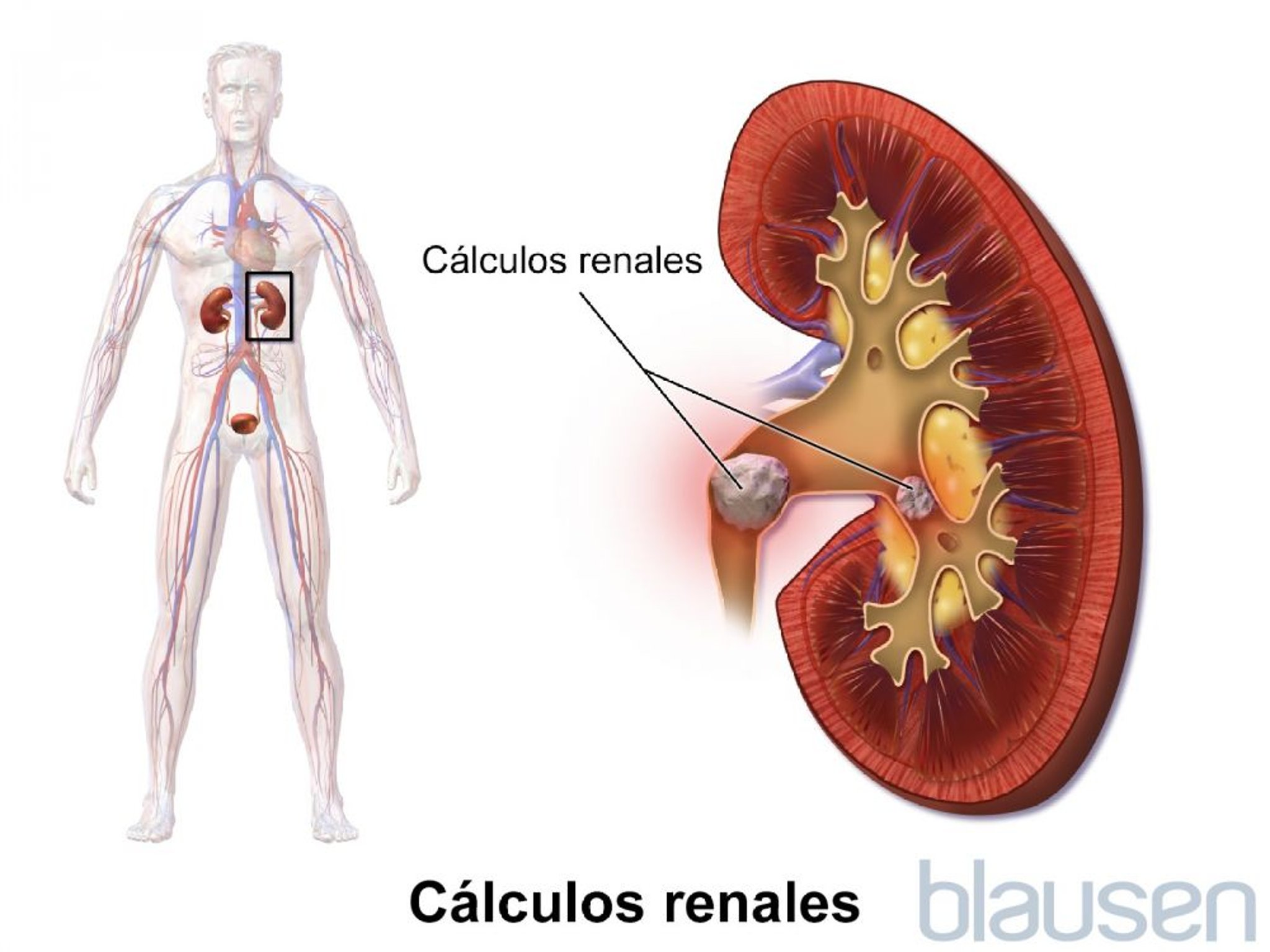
Además, los sujetos con este síndrome pueden desarrollar cálculos renales o una acumulación de calcio en los riñones (lo que se denomina nefrocalcinosis), ya que tienen grandes cantidades de calcio en la orina. No obstante, las personas con síndrome de Gitelman no desarrollan dichos problemas.
Diagnóstico del síndrome de Bartter y del síndrome de Gitelman
Determinación de las concentraciones de electrólitos en la sangre y en la orina
Los médicos sospechan el síndrome de Bartter o el síndrome de Gitelman en niños que presentan síntomas característicos o que tienen concentraciones anómalas de electrólitos en la sangre y la orina. A veces, los niveles anormales de electrolitos se detectan cuando se realizan pruebas de laboratorio por otras razones.
El diagnóstico del síndrome puede sugerirse mediante al encontrar altos niveles de renina y aldosterona en la sangre y altos niveles de sodio, cloruro y potasio en la orina.
El diagnóstico se confirma mediante pruebas genéticas, cada vez más disponibles.
Los familiares pueden ser evaluados por un especialista en genética (genetista) o un especialista en riñones (nefrólogo). Muy a menudo, se realizan pruebas genéticas.
Tratamiento del síndrome de Bartter y del síndrome de Gitelman
Complementos de sodio, potasio y magnesio
Para el síndrome de Bartter, fármacos antiinflamatorios no esteroideos
Debido a que no se puede corregir el déficit funcional de las células de los túbulos renales, el tratamiento es de por vida y está dirigido a corregir las anomalías hormonales e hidroelectrolíticas. Se añaden suplementos que contienen las sustancias que se pierden por la orina, como el sodio, el potasio y el magnesio, y también se aumenta la ingesta de líquidos.
Ciertos medicamentos pueden ser útiles. A las personas con el síndrome de Bartter se les prescriben fármacos antiinflamatorios no esteroideos (AINE), como la indometacina o ibuprofeno. Los AINE no son útiles en el síndrome de Gitelman.
Se puede administrar la hormona del crecimiento en niños de baja estatura.






