




Las hiperostosis craneotubulares son osteopetrosis que involucran crecimientos óseos excesivos que alteran el contorno y aumentan la densidad ósea.
Displasia diafisaria (enfermedad de Camurati-Engelmann)
Este trastorno autosómico dominante es causado por mutaciones en el gen TGFB1.
La displasia diafisaria se manifiesta en etapas medias de la infancia por dolor, debilidad y consunción muscular, en general de los miembros inferiores. Usualmente, estos síntomas se resuelven hacia los 30 años de edad. Las hiperostosis afectan a los huesos largos y el cráneo. En ocasiones, hay compresión de pares craneales y aumento de presión intracraneal. Algunos pacientes muestran discapacidad grave; otros son prácticamente asintomáticos.
El diagnóstico de la displasia diafisaria se sospecha por la combinación de déficits musculares y hiperostosis de los huesos largos y el cráneo. Por lo general, se realizan radiografías simples. El signo radiográfico predominante es el marcado engrosamiento de las superficies perióstica y medular de las corticales diafisarias de los huesos largos, pero los hallazgos son variables. Los conductos medulares y los contornos externos de los huesos son irregulares. Los miembros y el esqueleto axial suelen estar preservados. Rara vez, hay compromiso craneal, con ensanchamiento de la bóveda craneaal y esclerosis de la base del cráneo.
Los corticosteroides pueden ayudar a aliviar el dolor óseo y mejorar la fuerza muscular.
Hiperostosis endóstica (síndrome de van Buchem)
Por lo general, este trastorno es autosómico recesivo. En la hiperostosis endóstica, las lesiones genéticas parecen afectar la función normal de los osteoblastos.
El crecimiento excesivo y la distorsión de la mandíbula y la frente se tornan evidentes durante etapas medias de la infancia. Más adelante, hay atrapamiento de pares craneales, lo que provoca parálisis facial y sordera. No está afectada la expectativa de vida, la talla es normal y los huesos no son frágiles.
Las radiografías muestran ensanchamiento y esclerosis de la bóveda craneal, la base del cráneo y la mandíbula. El endostio diafisario de los huesos largos está engrosado.
La descompresión quirúrgica de los nervios atrapados puede ser útil.
Esclerosteosis
Este trastorno autosómico recesivo es causado por una mutación en el gen SOST, que codifica la proteína esclerostina. La esclerosteosis es más frecuente en los afrikáneres sudafricanos.
Durante la primera infancia, se observa crecimiento excesivo y esclerosis del esqueleto, en particular del cráneo. A menudo, la talla y el peso son excesivos. Los síntomas y los signos iniciales de la esclerosteosis pueden consistir en sordera y parálisis facial por atrapamiento de pares craneales. La distorsión de la cara, evidente a los 10 años de edad, termina siendo grave. La sindactilia cutánea u ósea del segundo y tercer dedo distingue la esclerosteosis de otras formas de hiperostosis craneotubulares.

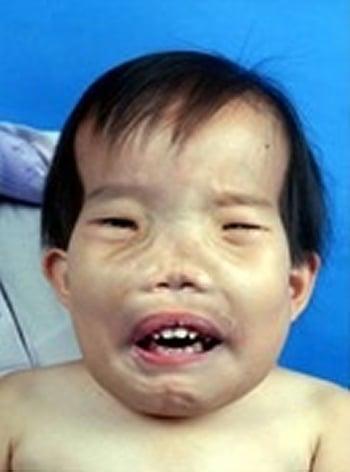
El diagnóstico de la esclerosteosis se sospecha por anomalías esqueléticas características, sobre todo cuando el paciente también tiene sindactilia. Por lo general, se realizan radiografías simples. Los signos radiológicos predominantes son ensanchamiento grosero y esclerosis de la bóveda craneal y la mandíbula. Hay preservación de los cuerpos vertebrales, aunque sus pedículos son densos. Los huesos de la pelvis muestran esclerosis, pero tienen contornos normales. Los huesos largos presentan corticales esclerosadas, hiperostóticas, y diáfisis poco modeladas. Existe una prueba genética diagnóstica para la confirmación.

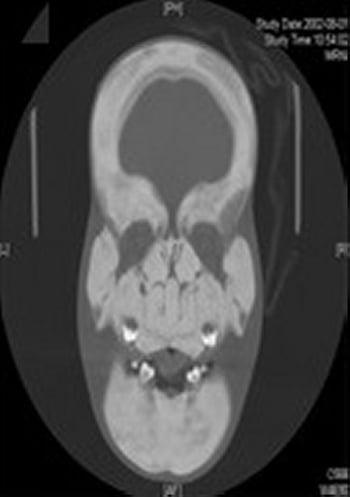
La cirugía para aliviar la presión intracraneana o descomprimir los nervios atrapados puede ser útil.









