




La sarcoidosis es un trastorno inflamatorio que produce granulomas no caseificantes en uno o más órganos y tejidos; se desconoce su etiología. Los pulmones y el sistema linfático son los más afectados, pero la sarcoidosis puede manifestarse en cualquier órgano. Los síntomas pulmonares varían desde ninguno hasta tos, disnea de esfuerzo y, rara vez, insuficiencia pulmonar o de otro órgano. El diagnóstico suele sospecharse primero por el compromiso pulmonar y se confirma por la radiografía de tórax, la biopsia y la exclusión de otras causas de inflamación granulomatosa. El tratamiento suele estar indicado en pacientes sintomáticos. El tratamiento de primera línea es con corticoides. El pronóstico es excelente para la enfermedad limitada, pero desfavorable para la más avanzada.
La sarcoidosis afecta con mayor frecuencia a personas de 20 a 40 años de edad, pero en ocasiones se presenta en niños y ancianos. A nivel mundial, la prevalencia es máxima en estadounidenses de raza negra y en etnias del norte de Europa, en especial en escandinavos. La presentación de la enfermedad tiene amplias variaciones según los antecedentes raciales y étnicos; los estadounidenses de raza negra tienen con mayor frecuencia manifestaciones extratorácicas. La sarcoidosis es un poco más frecuente en las mujeres.
Síndrome de Löfgren
El síndrome de Löfgren se manifiesta como una tríada de poliartritis migratoria aguda, eritema nodoso y adenopatías hiliares. También puede haber fiebre, malestar general, uveítis y parotiditis. El síndrome de Löfgren es más común entre las personas de ascendencia europea. El síndrome de Löfgren es autolimitado. Por lo general, los pacientes pueden tratarse solo con antiinflamatorios no esteroideos (AINE). La tasa de recidivas es baja.
Síndrome de Heerfordt
El síndrome de Heerfordt (fiebre uveoparotídea) se manifiesta con tumefacción de la glándula parótida (debido a infiltrado sarcoideo), uveítis, fiebre crónica y, con menor frecuencia, parálisis del nervio facial. El síndrome de Heerfordt puede ser autolimitado. El tratamiento es similar al de la sarcoidosis.
Síndrome de Blau
El síndrome de Blau es una enfermedad semejante a la sarcoidosis heredada de forma autosómica dominante que se manifiesta en los niños. No se sabe si el síndrome de Blau surge a través del mismo mecanismo que la sarcoidosis diagnosticada en adultos. En el síndrome de Blau, los niños se presentan antes de la edad de 4 años, con artritis, erupciones cutáneas, y uveítis. El síndrome de Blau es usualmente autolimitado. Los síntomas se suelen aliviar con medicamentos antiinflamatorios no esteroideos.
Etiología de la sarcoidosis
Se considera que la sarcoidosis se debe a una respuesta inflamatoria exagerada ante un antígeno del medioambiente en una persona genéticamente susceptible. Los desencadenantes propuestos son
Propionibacterium acnes y micobacterias (potencialmente la proteína catalasa-peroxidasa [mKatG] de Mycobacterium tuberculosis)
El moho y ciertas sustancias no identificadas presentes en los lugares de trabajo con olor a humedad
Pesticidas, en particular los que contienen compuestos de aluminio
El consumo de tabaco se correlaciona inversamente con la sarcoidosis.
La evidencia que apoya la susceptibilidad genética incluye lo siguiente:
Mayor tasa de concordancia de la enfermedad en gemelos monocigóticos que en gemelos dicigóticos
El aumento de la prevalencia de la sarcoidosis (alrededor de 3,6 a 9,6%) entre los familiares de primero o segundo grado de pacientes que tienen sarcoidosis
Aumento de cinco veces en el riesgo relativo de desarrollar sarcoidosis en hermanos de pacientes que tienen sarcoidosis
Identificación de varios genes del antígeno leucocitario humano (HLA, human leukocyte antigen) y no HLA posiblemente asociados con el riesgo, la evolución y los fenotipos de la sarcoidosis
Por ejemplo, el haplotipo HLA-DRB1*03/DQB1*02 se asocia con el síndrome de Löfgren y predice un pronóstico excelente, a diferencia del HLA-DRB1*15/HLA DQB1*0602, que predice la persistencia de la enfermedad.
Fisiopatología de la sarcoidosis
El antígeno desconocido desencadena una respuesta inmunitaria mediada por células que se caracteriza por la acumulación de células T y macrófagos, la liberación de citocinas y quimiocinas y la organización de las células respondedoras en granulomas. La agrupación de la enfermedad en familias y comunidades sugiere una predisposición genética, exposiciones compartidas o transmisión interpersonal, menos probable.
El proceso inflamatorio lleva a la formación de granulomas no caseificantes, la característica anatomopatológica distintiva de la sarcoidosis. Los granulomas son colecciones de células mononucleares y macrófagos que se diferencian en células gigantes multinucleadas y epitelioides, y están rodeadas por linfocitos, plasmocitos, fibroblastos y colágeno. Los granulomas se producen con mayor frecuencia en los pulmones y los ganglios linfáticos, pero pueden afectar a cualquier órgano y causar disfunción significativa. Los granulomas en los pulmones se distribuyen a lo largo de los vasos linfáticos, la mayoría en las regiones peribronquiolar, subpleurales y perilobulillares. La acumulación de granulomas distorsiona la estructura de los órganos afectados. No se sabe si los granulomas producen fibrosis en forma directa o se desarrollan en paralelo.
Puede ocurrir hipercalcemia debido al aumento de la conversión de la vitamina D a la forma activada (1,25 hidroxivitamina D) por los macrófagos. La hipercalciuria puede estar presente, incluso en pacientes con niveles normales de calcio en suero. Pueden ocurrir nefrolitiasis y nefrocalcinosis, que a veces llevan a la enfermedad renal crónica.

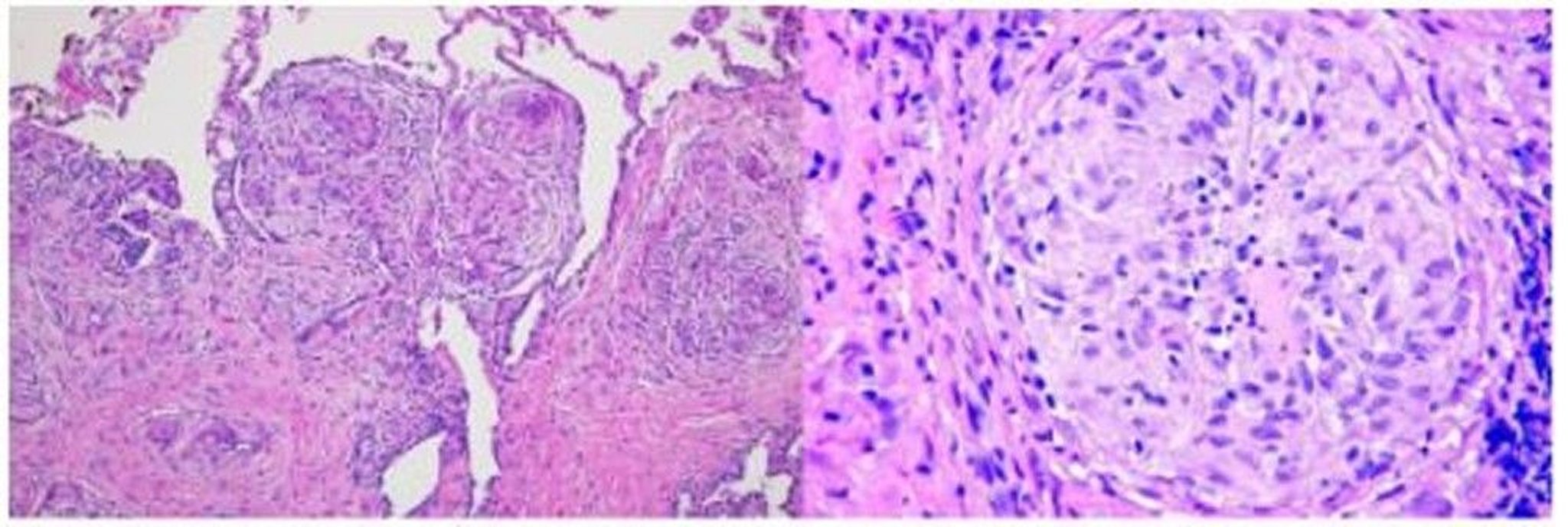
Imagen cortesía de Birendra P. Sah, MD, FCCP.
Signos y síntomas de la sarcoidosis
Los signos y síntomas dependen del sitio y el grado de implicación y varían con el tiempo, que van desde la remisión espontánea a la enfermedad crónica poco activa. En consecuencia, se necesita una revaluación frecuente para identificar nuevos síntomas y el compromiso de diferentes órganos. La mayoría de los casos es probablemente asintomática y, por lo tanto, no es detectada. La enfermedad pulmonar aparece en > 90% de los pacientes adultos.
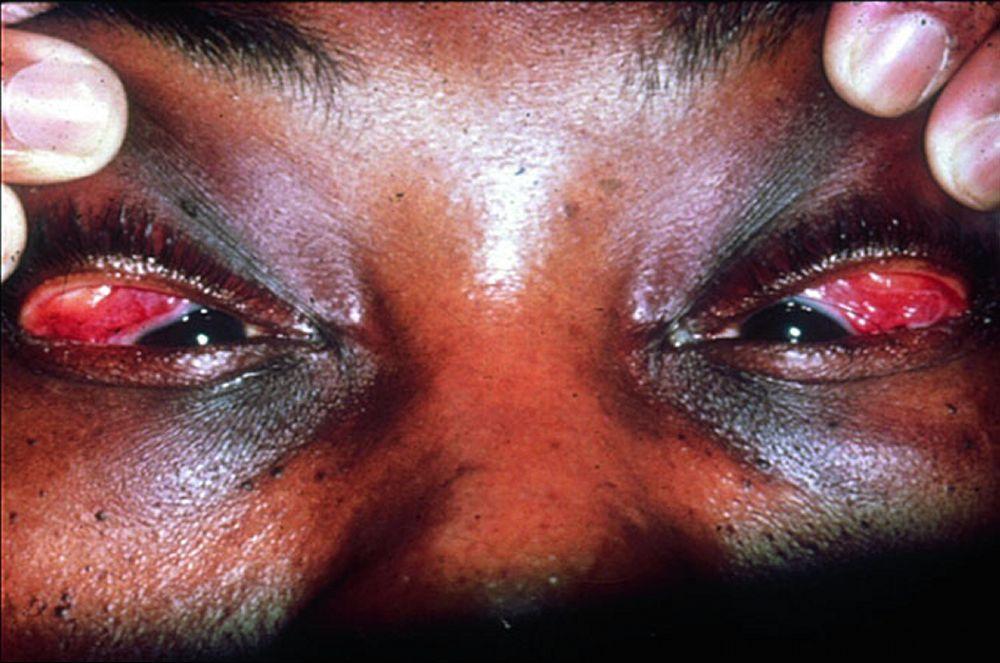
Los signos y síntomas pueden incluir disnea, tos, malestar en el pecho y crepitantes. También son comunes la astenia, el malestar general, la debilidad, la anorexia, la pérdida de peso y la febrícula. La sarcoidosis se puede manifestar como fiebre de etiología desconocida. El compromiso sistémico provoca diversos síntomas (véase tabla Compromiso sistémico en la sarcoidosis), que varían según la raza, el sexo y la edad. Los pacientes de raza negra tienen más probabilidades que los de raza blanca a presentar compromiso de los ojos, el hígado, la médula ósea, los ganglios linfáticos periféricos y la piel; el eritema nudoso es una excepción. Las mujeres son más propensas a tener eritema nudoso y compromiso ocular o del sistema nervioso. Los varones y los pacientes de edad avanzada están más propensos a presentar hipercalcemia.
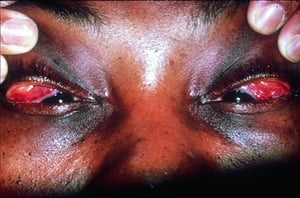
© Springer Science+Business Media
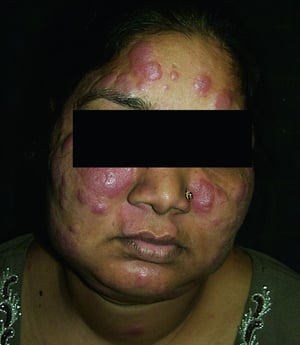
© Springer Science+Business Media
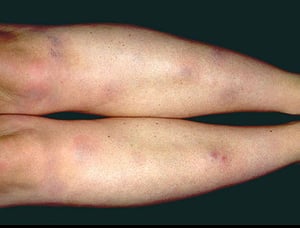
Imagen cortesía de Thomas Habif, MD.
© Springer Science+Business Media
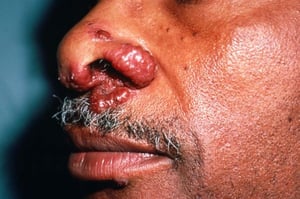
Imagen cortesía de Karen McKoy, MD.
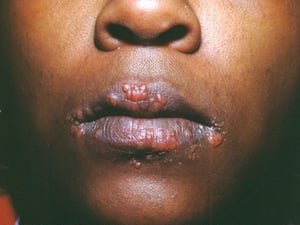
Imagen cortesía de Karen McKoy, MD.
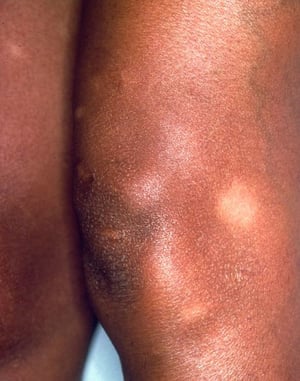
Imagen cortesía de Karen McKoy, MD.

© Springer Science+Business Media

© Springer Science+Business Media

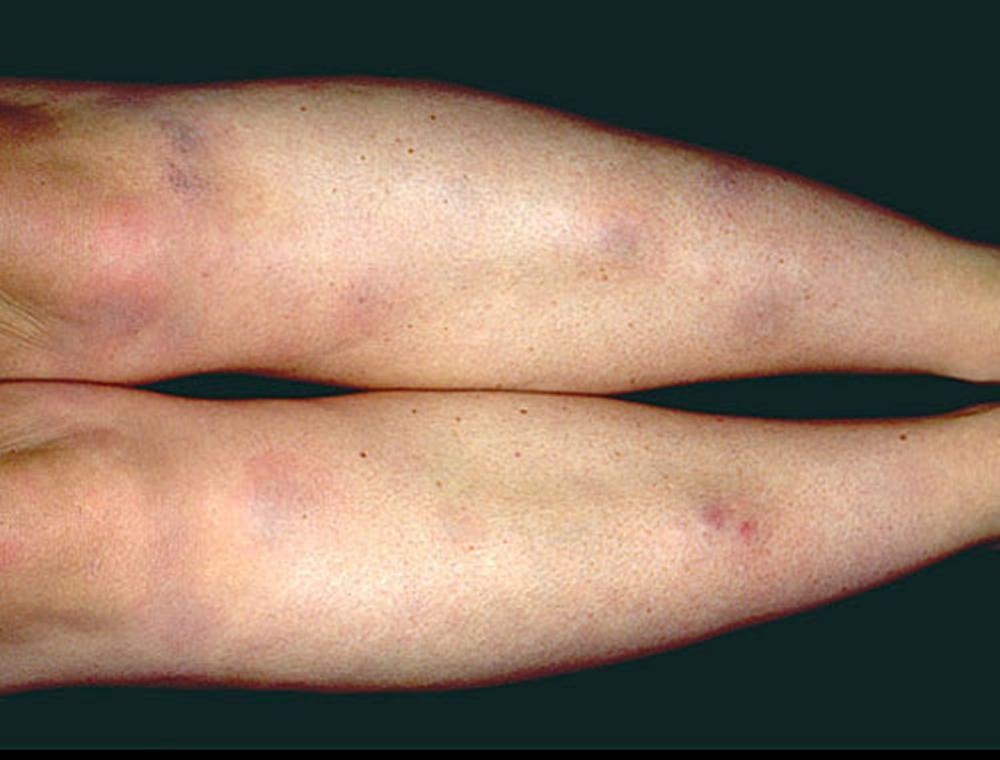
Imagen cortesía de Thomas Habif, MD.

© Springer Science+Business Media

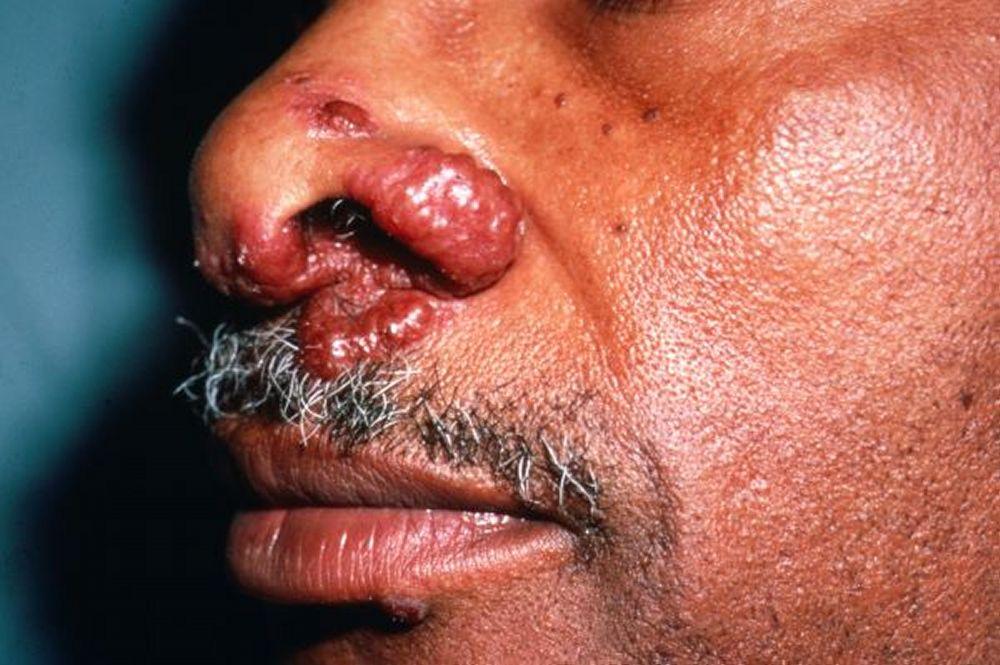
Imagen cortesía de Karen McKoy, MD.

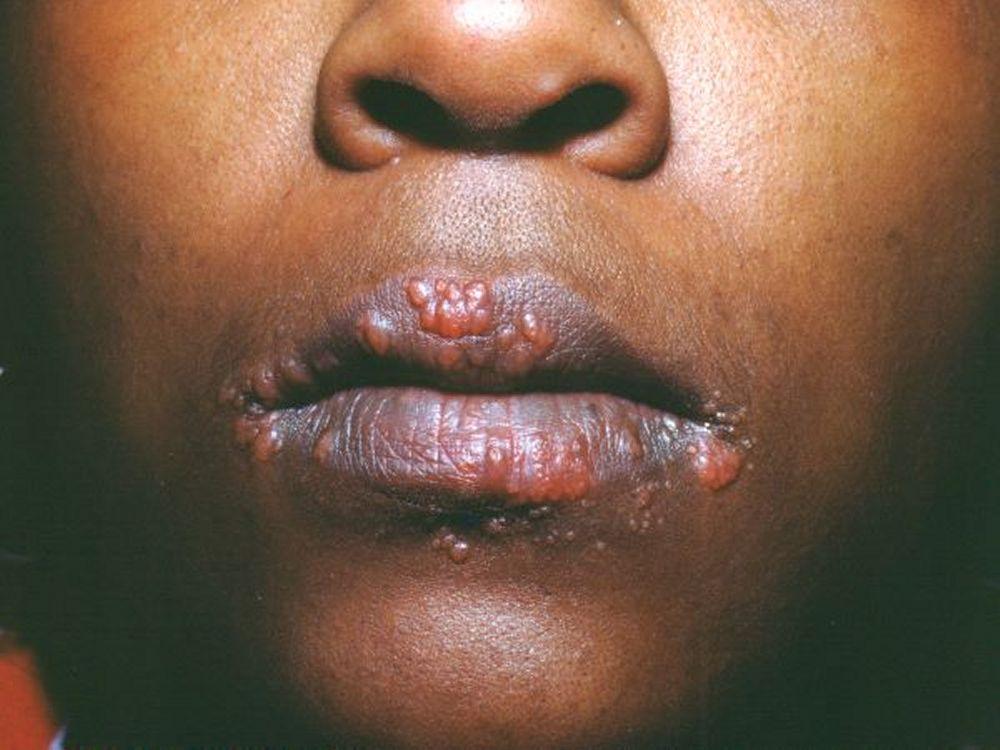
Imagen cortesía de Karen McKoy, MD.

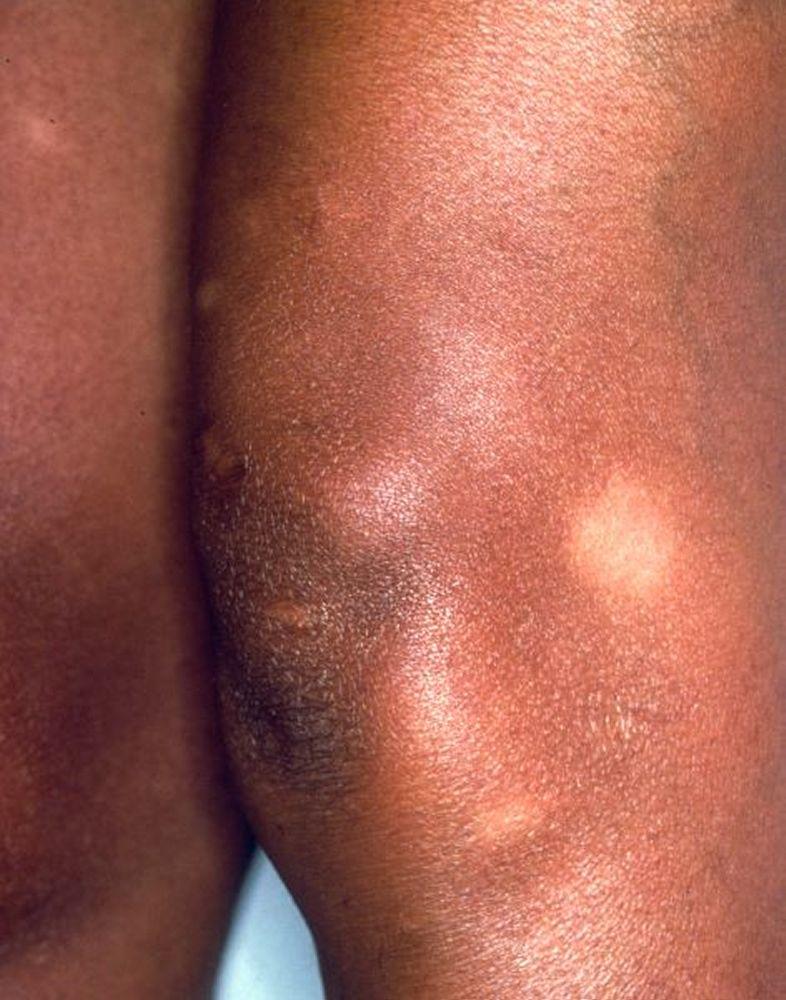
Imagen cortesía de Karen McKoy, MD.
Los niños con sarcoidosis pueden presentar el síndrome de Blau (artritis, erupción cutánea, uveítis), o manifestaciones similares a las de los adultos. En este grupo etario, la sarcoidosis puede confundirse con artritis juvenil idiopática (artritis reumatoide juvenil).
Diagnóstico de la sarcoidosis
Estudios de diagnóstico por imágenes del tórax
Biopsia
Exclusión de otros trastornos granulomatosos
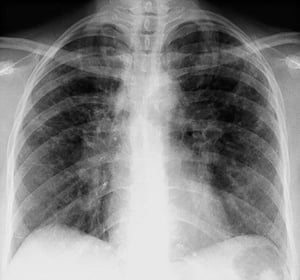
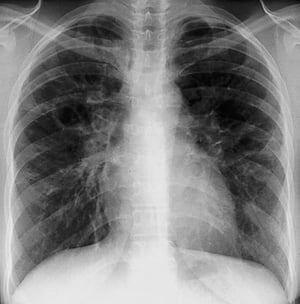
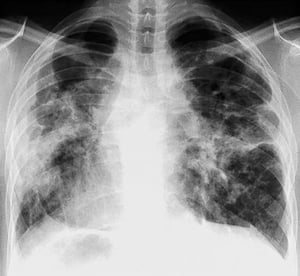
A menudo, la sarcoidosis se sospecha cuando se detecta en forma accidental adenopatías hiliares con o sin infiltrados pulmonares, en una radiografía de tórax. La adenopatía hiliar bilateral es la anormalidad más común.
Si se sospecha sarcoidosis, la radiografía de tórax debe ser la primera prueba si ya no ha sido hecha. El aspecto radiográfico tiende a predecir bastante la probabilidad de remisión espontánea (véase tabla Radiografía de tórax en la estadificación de la Sarcoidosis) en pacientes con compromiso aislado de los ganglios linfáticos torácicos. Sin embargo, estadificar la sarcoidosis por la radiografía de tórax puede ser engañoso; por ejemplo, la sarcoidosis extrapulmonar, al igual que la sarcoidosis cardiaca o neurológica, puede presagiar un pronóstico en ausencia de afectación pulmonar. Además, los hallazgos en las radiografías de tórax predicen mal la función pulmonar, por lo que la apariencia radiológica puede no indicar con precisión la gravedad de la sarcoidosis pulmonar.
Una radiografía de tórax normal (fase 0) no excluye el diagnóstico de sarcoidosis, sobre todo cuando se sospecha de afectación cardíaca o neurológica. Una tomografía computarizada de alta resolución es más sensible para la detección de adenopatías hiliares y mediastínicas y anormalidades del parénquima. El compromiso del parénquima pulmonar predomina en los lóbulos superiores, pero puede observarse en cualquier parte de los pulmones. Los hallazgos tomográficos (véase también imagen TC de tórax en la sarcoidosis pulmonar) en estadios más avanzados (II a IV) incluyen los siguientes:
Engrosamiento de los haces broncovasculares y las paredes bronquiales
Reborde de los tabiques interlobulillares
Opacificación en vidrio esmerilado
Nódulos parenquimatosos, quistes o cavidades
Bronquiectasias por tracción
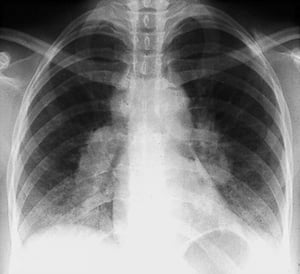
Con autorización del editor. De Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Publicado por J Crapo. Philadelphia, Current Medicine, 2005.
Con autorización del editor. De Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Publicado por J Crapo. Philadelphia, Current Medicine, 2005.
Con autorización del editor. De Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Publicado por J Crapo. Philadelphia, Current Medicine, 2005.
Con autorización del editor. De Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Publicado por J Crapo. Philadelphia, Current Medicine, 2005.
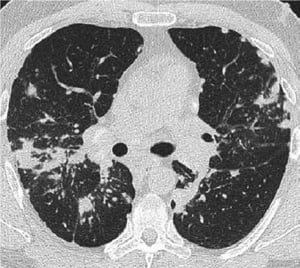
Imagen cortesía de Birendra P. Sah, MD, FCCP.

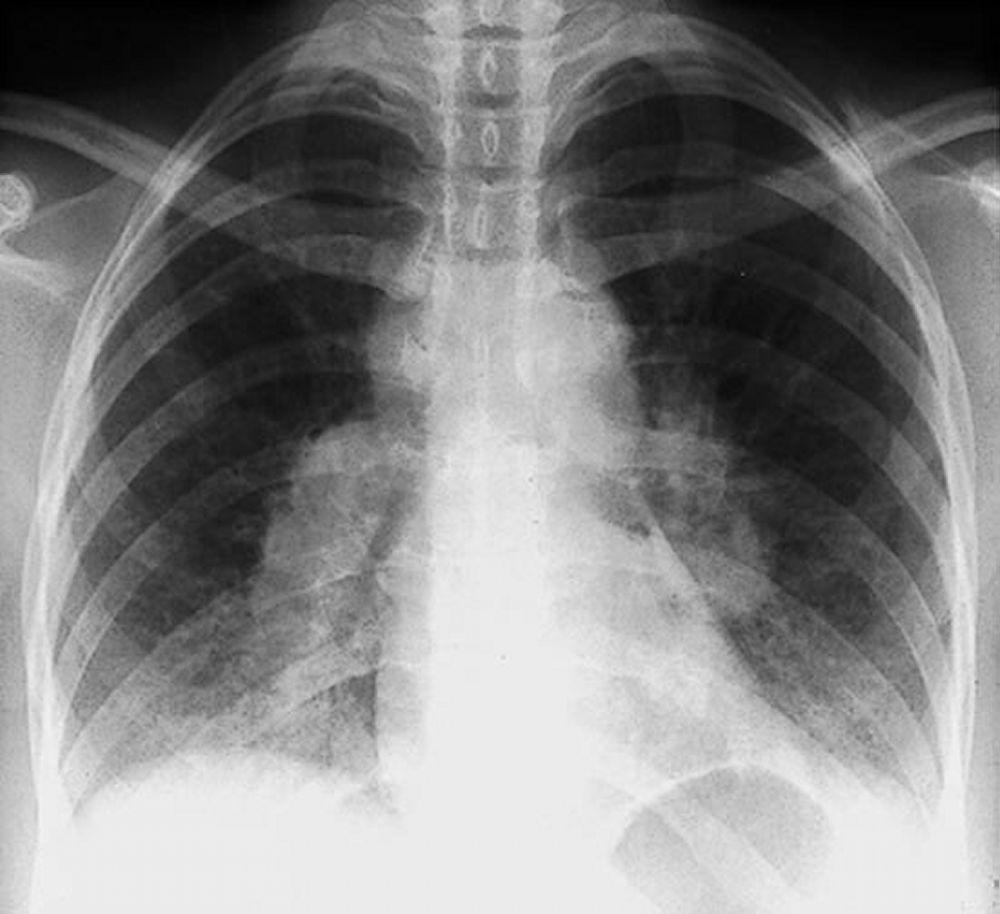
Con autorización del editor. De Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Publicado por J Crapo. Philadelphia, Current Medicine, 2005.

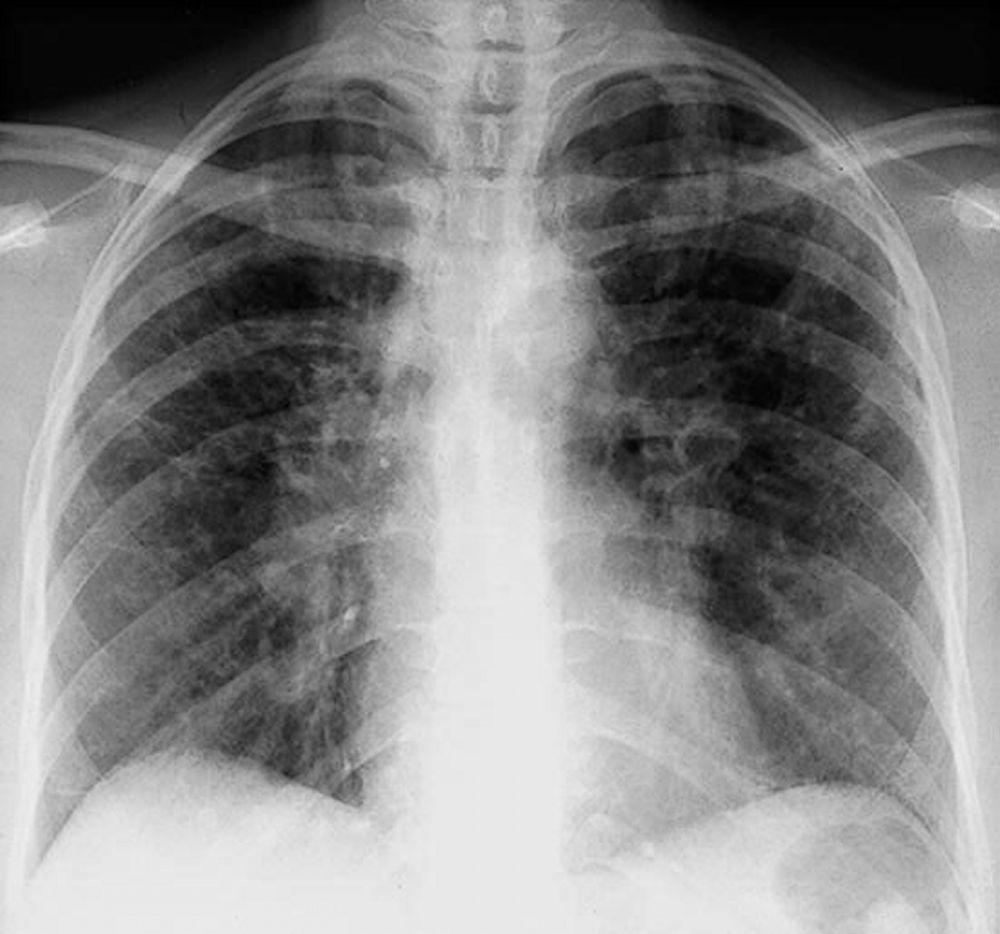
Con autorización del editor. De Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Publicado por J Crapo. Philadelphia, Current Medicine, 2005.

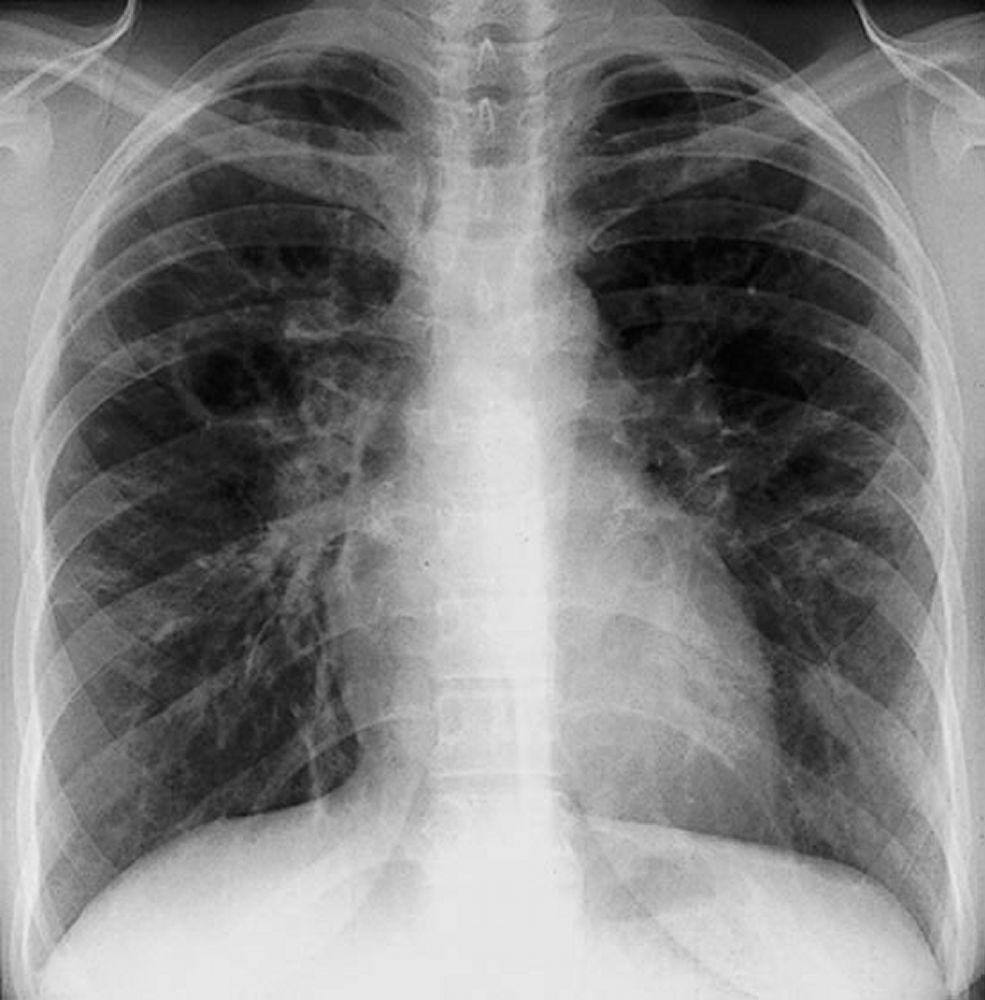
Con autorización del editor. De Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Publicado por J Crapo. Philadelphia, Current Medicine, 2005.

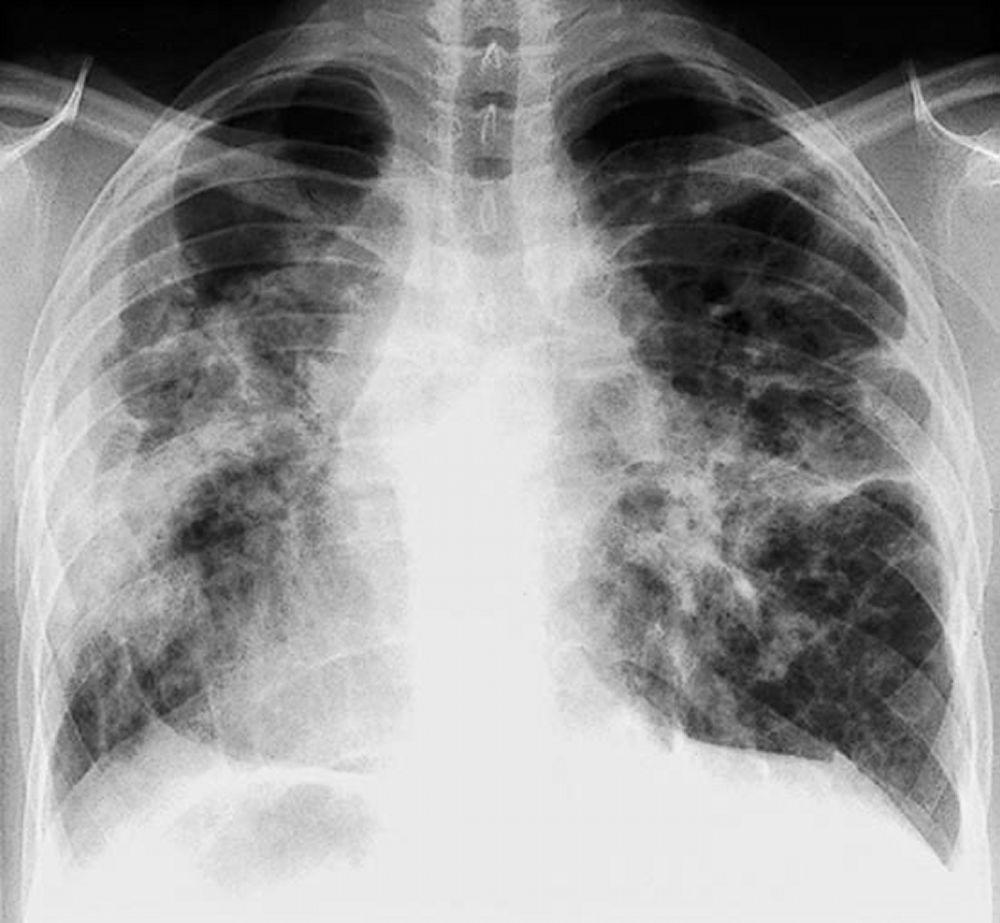
Con autorización del editor. De Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Publicado por J Crapo. Philadelphia, Current Medicine, 2005.

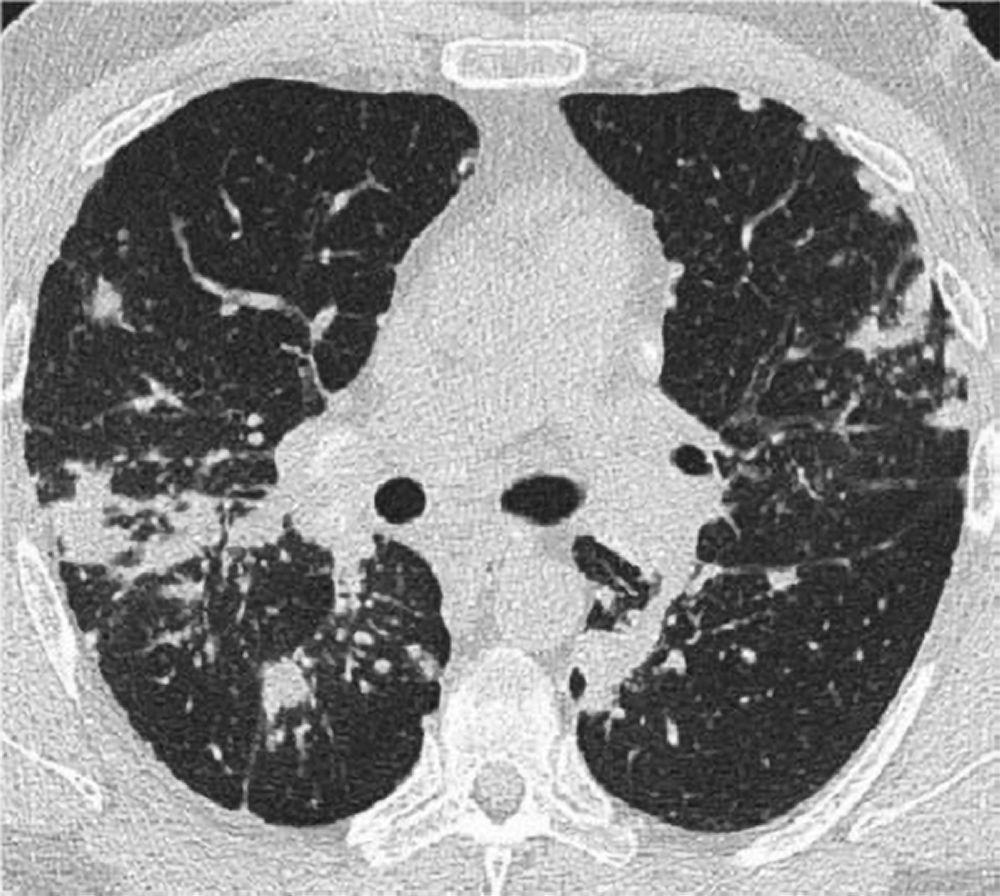
Imagen cortesía de Birendra P. Sah, MD, FCCP.
Cuando los estudios por la imagen sugieren sarcoidosis, el diagnóstico se confirma por la demostración de granulomas no caseificantes en la biopsia y la exclusión de otras causas de enfermedad granulomatosa (véase tabla Diagnósticos diferenciales de la sarcoidosis). El síndrome de Löfgren no requiere confirmación por biopsia.
Por consiguiente, para la evaluación diagnóstica se necesita:
Selección del sitio de biopsia
Exclusión de otras causas de enfermedad granulomatosa
Evaluación de la gravedad y la extensión de la enfermedad para determinar si está indicado el tratamiento
Sitios para biopsia
Los sitios adecuados para la biopsia pueden ser evidentes por el examen físico y la evaluación inicial; los ganglios linfáticos periféricos, las lesiones de la piel y la conjuntiva son de fácil acceso. La aspiración transbronquial con aguja guiada por ecografía endobronquial de un ganglio linfático del mediastino o el hilio tiene un rendimiento diagnóstico informado de alrededor del 90% y es el procedimiento diagnóstico de elección en pacientes con afectación intratorácica.
Cuando se sospecha sarcoidosis pulmonar y la punción guiada por ecografía endobronquial no es diagnóstica, se puede efectuar una biopsia pulmonar transbronquial broncoscópica con lavado broncoalveolar (LBA). También se puede utilizar en pacientes sin infiltrados en el parénquima pulmonar porque el rendimiento diagnóstico de la biopsia pulmonar transbronquial en la sarcoidosis estadio I es de alrededor del 50%. Si la biopsia transbronquial por broncoscopia no es diagnóstica, se puede intentar por segunda vez.
Si la punción guiada por ecografía endobronquial y la biopsia transbronquial por broncoscopia no son diagnósticas o si la broncoscopia no se puede tolerar, se puede hacer mediastinoscopia para biopsiar los ganglios linfáticos mediastínicos o hiliares, o biopsia de pulmón asistida por videotoracoscopía o biopsia pulmonar a cielo abierto, para obtener tejido pulmonar. Si la sarcoidosis se sospecha fuertemente pero no es evidente en el sitio de biopsia en base a los hallazgos del examen o de las imágenes, la tomografía por emisión de positrones (PET) puede ayudar a identificar sitios activos ocultos, tales como el corazón, el hueso, el músculo y el cerebro.
Exclusión de otros diagnósticos
Es fundamental descartar otros diagnósticos, sobre todo cuando los signos y síntomas radiográficos son mínimos, porque muchos otros trastornos y procesos pueden causar inflamación granulomatosa (véase tabla Diagnósticos diferenciales de la sarcoidosis). El tejido de biopsia debe ser cultivado para hongos y micobacterias. Deben investigarse los antecedentes de exposición ocupacional (p. ej., silicatos, berilio), medioambientales (p. ej., heno mohoso, aves y otros desencadenantes antigénicos de neumonitis por hipersensibilidad) y los antígenos infecciosos (p. ej., tuberculosis, coccidioidomicosis, histoplasmosis). Las pruebas cutáneas de derivados proteicos purificados (PPD) o el ensayo de liberación de interferón gamma deben realizarse al principio de la evaluación.
Evaluación de la gravedad de la enfermedad
La gravedad se evalúa de acuerdo con el compromiso del órgano, como, por ejemplo, solo con afectación pulmonar
Pruebas de la función pulmonar
Los resultados de las pruebas de la función pulmonar a menudo son normales en los estadios tempranos, pero demuestran restricción y reducción de la capacidad de difusión del monóxido de carbono (DLCO) en la enfermedad avanzada. La obstrucción del flujo de aire también se produce y puede sugerir la participación de la mucosa bronquial. El agregado de una prueba de caminata de 6 minutos puede caracterizar el deterioro funcional de manera más completa que los resultados aislados de las pruebas de función pulmonar. Los pacientes con compromiso pulmonar extenso pueden tener saturación de oxígeno normal en reposo, pero pueden mostrar desaturación con el esfuerzo.
Las pruebas de cribado de rutina recomendadas para la enfermedad extrapulmonar son
ECG de 12 derivaciones y ecocardiografía
Examen oftálmico con lámpara de hendidura
Pruebas habituales en sangre para evaluar la función renal y hepática
Niveles séricos de calcio y excreción de calcio en orina de 24 horas
A menudo se requieren estudios de diagnóstico por imágenes para detectar la sarcoidosis extrapulmonar. La resonancia magnética cardíaca (RM) con y sin contraste de gadolinio puede ser apropiada en pacientes con síntomas cardíacos. En pacientes con síntomas neurológicos, puede ser necesaria una RM de cerebro o de la columna vertebral con o sin gadolinio. Las gammagrafías óseas y la electromiografía pueden ser apropiadas en pacientes con síntomas reumatológicos. La TC con emisión de positrones parece ser la prueba más sensible para la detección de sarcoidosis ósea y otras localizaciones extrapulmonares y se utiliza junto con la RM en pacientes con compromiso cardíaco. La TC abdominal con sustancias radiopacas no se recomienda en forma sistemática, pero puede proporcionar evidencia de compromiso hepático o esplénico (p. ej., aumento de tamaño, lesiones hipolúcidas). La gammagrafía de cuerpo entero con galio se ha sustituido en gran medida por la TC con emisión de positrones. Si está disponible, la gammagrafía con galio puede proporcionar evidencia de apoyo útil en ausencia de confirmación tisular. El aumento simétrico de la captación en ganglios mediastínicos e hiliares (signo lambda) y en las glándulas lagrimales, las glándulas parótidas y salivales (signo del panda) sugiere fuertemente una sarcoidosis. Un resultado negativo en pacientes que reciben prednisona no es fiable.
Las pruebas de laboratorio desempeñan un papel adyuvante para establecer el diagnóstico y determinar el grado de compromiso de los órganos. El hemograma completo con recuento diferencial puede mostrar anemia, eosinofilia o leucopenia. Debe medirse el calcio en suero para detectar hipercalcemia. Los resultados de las pruebas para nitrógeno ureico en sangre, creatinina y de la función hepática pueden estar elevadas en la sarcoidosis renal y hepática. La proteína total puede estar elevada debido a la hipergammaglobulinemia. El aumento de la velocidad de eritrosedimentación es común, pero inespecífico. Se recomienda medir el nivel de calcio en una muestra de orina recogida durante 24 horas para descartar hipercalciuria, incluso en pacientes con valores normales de calcemia. Las concentraciones séricas elevadas de la enzima convertidora de la angiotensina (ECA) sugieren sarcoidosis, pero son inespecíficas y pueden estar aumentadas en pacientes con otras enfermedades (p. ej., hipertiroidismo, diabetes, enfermedad de Gaucher, silicosis, enfermedad por micobacterias, infecciones micóticas, neumonitis por hipersensibilidad, linfoma). Los niveles de enzima convertidora de angiotensina (ECA), si están elevados, pueden ser útiles para controlar el cumplimiento del tratamiento con corticosteroides. Los niveles de ECA descienden, incluso cuando los pacientes reciben dosis bajas de corticosteroides.
Debe realizarse un lavado broncoalveolar junto con la biopsia broncoscópica para descartar posibles infecciones (p. ej., cuando los hallazgos con medios de diagnóstico menos invasivos como los estudios de diagnóstico por imágenes no son típicos de la sarcoidosis) y para excluir otras formas de enfermedad pulmonar intersticial si el diagnóstico de sarcoidosis está en duda. Los hallazgos del lavado broncoalveolar presentan amplias variaciones, pero la linfocitosis (linfocitos > 15%) o una relación CD4+/CD8+> 3,5 en el recuento diferencial de células en el líquido de lavado sugieren el diagnóstico en el contexto clínico adecuado. Sin embargo, la ausencia de estos hallazgos no descarta sarcoidosis.
Tratamiento de la sarcoidosis
Medicamentos antiinflamatorios no esteroideos
Corticosteroides
Inmunosupresores
Anticuerpos anti-factor de necrosis tumoral alfa
Dado que la sarcoidosis a menudo resuelve espontáneamente, los pacientes con síntomas leves y los que son asintomáticos no requieren tratamiento, aunque deben ser controlados en busca de signos de deterioro. El seguimiento de estos pacientes puede realizarse con radiografías seriadas de tórax, pruebas de la función pulmonar (que incluyen la capacidad de difusión) y marcadores de compromiso extratorácico (p. ej., pruebas habituales de la función renal y hepática, examen oftalmológico anual con lámpara de hendidura). La frecuencia de las pruebas de seguimiento se determina de acuerdo con la gravedad de la enfermedad.
Los pacientes que requieren tratamiento en forma independiente del estadio de la radiografía de tórax incluyen aquellos con los siguientes:
Síntomas de empeoramiento
Limitación de la actividad
Función pulmonar marcadamente anormal o con deterioro progresivo
Alteraciones radiográficas preocupantes (p. ej., cavitación, fibrosis, masas conglomeradas, signos de hipertensión pulmonar)
Compromiso cardíaco, del sistema nervioso u ocular
Insuficiencia renal o hepática
Hipercalcemia moderada a grave
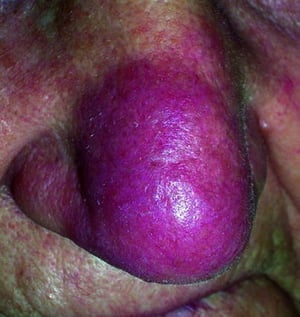
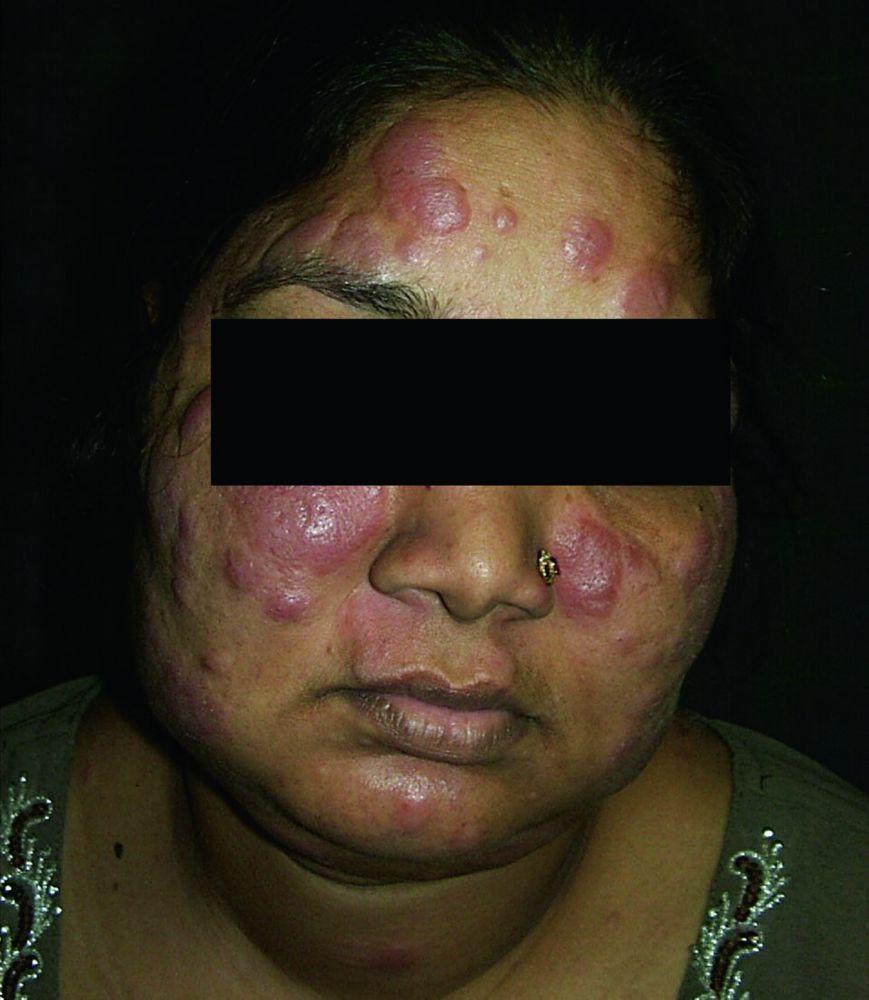
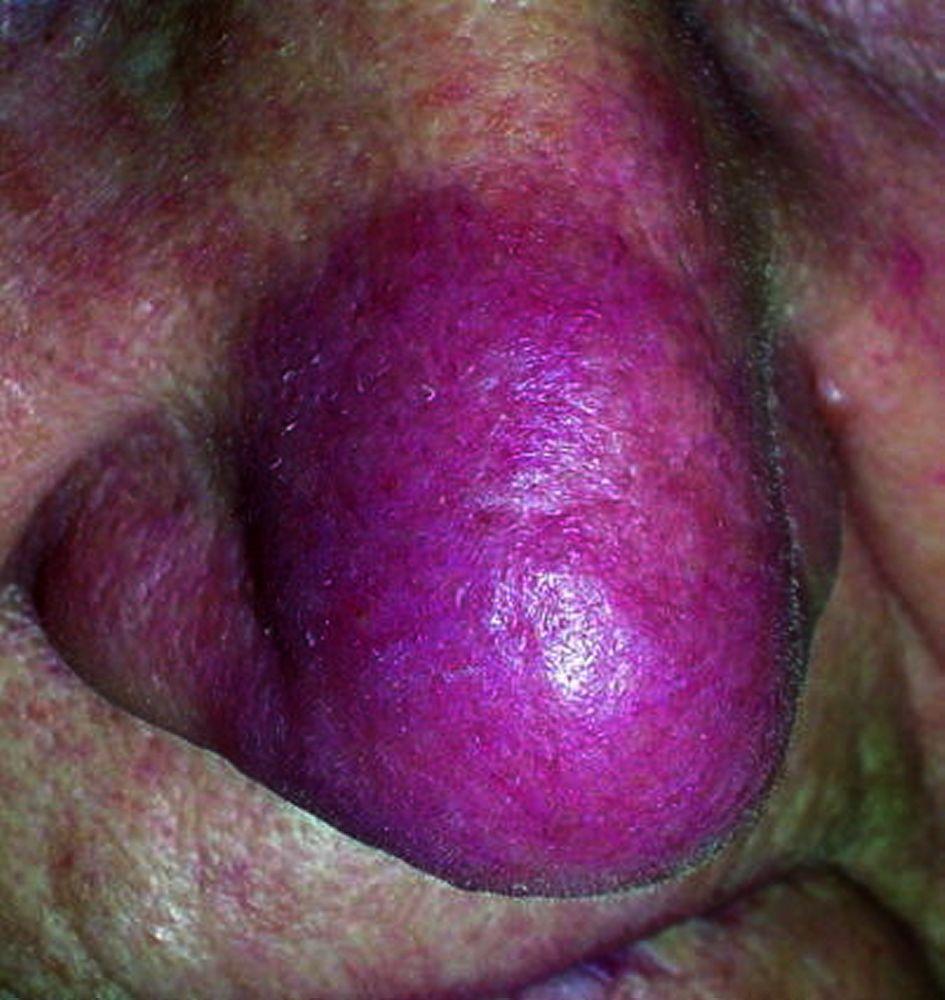
Enfermedad desfigurante cutánea (p. ej., lupus pernio) o articular
Se utilizan medicamentos antiinflamatorios no esteroideos para tratar el malestar musculoesquelético.
Corticosteroides
El manejo de los síntomas se inicia con corticosteroides. La presencia de anomalías en la imagen del tórax sin síntomas significativos o evidencia de disminución de la función del órgano no es una indicación para el tratamiento. Un protocolo estándar consiste en prednisona, de 20 a 40 mg por vía oral 1 vez al día, dependiendo de los síntomas y la gravedad de los hallazgos. También se pueden usar regímenes con días alternados: p. ej., prednisona, 40 mg por vía oral 1 vez en días alternos. Aunque los pacientes rara vez requieren > 40 mg/día, dosis más altas pueden necesitarse para reducir las complicaciones en la enfermedad neurológica grave y cardíaca. La respuesta suele producirse en el transcurso de 6 a 12 semanas, por lo que los síntomas, otros marcadores de gravedad de la enfermedad y las pruebas de la función pulmonar pueden volver a evaluarse en 6 a 12 semanas. Los casos insidiosos y crónicos pueden responder más lentamente. Los corticosteroides se disminuyen en forma gradual hasta la dosis de mantenimiento (p. ej., prednisona 10 a 15 mg/día) después de la evidencia de respuesta y se continúan durante un períod adicional de 6 a 9 meses en caso de mejoría.
Se desconoce la duración óptima del tratamiento. Las reducciones prematuras pueden dar por resultado la aparición de recidivas. El fármaco se interrumpe lentamente si no hay respuesta o ésta es ambigua. Por último, los corticoides pueden interrumpirse en la mayoría de los pacientes, pero como la recidiva se produce en hasta el 50% de las veces, debe repetirse la monitorización en general a los 3 a 6 meses. El tratamiento con corticosteroides debe reanudarse si recidivan los signos y los síntomas. Debido a que la producción de la enzima convertidora de angiotensina (ECA) se suprime con dosis bajas de corticosteroides, la medición seriada de sus niveles séricos puede ser útil en la evaluación del cumplimiento del tratamiento con corticosteroides en pacientes que tienen niveles elevados de la ECA.
Los corticosteroides inhalados pueden aliviar la tos en pacientes con afectación endobronquial. Se puede agregar un broncodilatador inhalatorio en pacientes con enfermedad obstructiva de las vías aéreas.
Los corticoides tópicos pueden ser útiles en algunos casos de enfermedad cutánea, de los senos nasales y ocular.
Se recomienda la profilaxis contra la neumonía por Pneumocystis jirovecii mientras los pacientes reciben > 20 mg de prednisona 1 vez al día o su equivalente durante más de un mes y en los que reciben inmunosupresores.
El alendronato u otro bisfosfonato puede ser el tratamiento de elección para la prevención de la osteoporosis inducida por corticosteroides en personas en riesgo (p. ej., pacientes mayores). El uso de suplementos de calcio o vitamina D puede ocasionar hipercalcemia debido a la producción endógena de vitamina D activa (1, 25 dihidroxivitamina D) por los granulomas sarcoidales. Las mediciones de calcio en suero y en orina de 24 horas deben ser normales antes de administrar estos suplementos.
Perlas y errores
|
Inmunosupresores
Los inmunosupresores se utilizan cuando
Los pacientes no toleran la prednisona
La sarcoidosis es refractaria a dosis moderadas a altas de prednisona
La dosis de prednisona no puede reducirse por debajo de 10 a 15 mg diarios después de 3 meses
Antes de agregar otros inmunosupresores, se deben considerar las posibles razones de la falta de mejoría clínica, como el incumplimiento, la enfermedad comórbida (p. ej., asma, insuficiencia cardíaca, anemia), hipertensión pulmonar, y la fibrosis terminal.
El metotrexato es el inmunosupresor usado con mayor frecuencia. Los pacientes deben someterse a una prueba de metotrexato en dosis de 10 a 15 mg/semana durante 6 meses. Antes de comenzar con el metotrexato, los pacientes deben ser evaluados para detectar una infección por el virus de la hepatitis B y el virus de la hepatitis C. Inicialmente, se administran metotrexato y corticoides; a las 6 a 8 semanas, la dosis de corticoides puede reducirse y, en muchos casos, interrumpirse. La respuesta máxima al metotrexato, sin embargo, puede insumir de 6 a 12 meses. En estos casos, la prednisona debe reducirse más lentamente. Los recuentos seriados de sangre y las pruebas de enzimas hepáticas deben hacerse cada 2 a 4 semanas al principio y después cada 6 a 12 semanas una vez lograda una dosis estable. Se recomienda la administración de ácido fólico (1 mg por vía oral una vez al día) para los pacientes tratados con metotrexato con el fin de reducir el riesgo de efectos adversos.
Otros inmunosupresores incluyen azatioprina, micofenolato, ciclofosfamida, leflunomida e hidroxicloroquina. La hidroxicloroquina, 400 mg por vía oral 1 vez al día o 200 mg por vía oral 2 veces al día puede ser eficaz para tratar la hipercalcemia, artralgias, la sarcoidosis cutánea o los ganglios linfáticos periféricos muy aumentados de tamaño que provocan malestar o desfiguración. La evaluación oftalmológica debe realizarse antes de iniciar la hidroxicloroquina y cada 6 a 12 meses durante el tratamiento para controlar su toxicidad ocular.
La recidiva es frecuente una vez que se suspende un inmunosupresor.
Anticuerpos anti-factor de necrosis tumoral alfa
El infliximab generalmente se usa como medicamento de tercera línea para tratar la sarcoidosis refractaria y para los pacientes que no toleran los corticosteroides y los inmunosupresores ya mencionados. Antes de comenzar la terapia, los pacientes deben someterse a una prueba con un derivado proteico purificado (PPD) o a un ensayo de liberación de interferón gamma para detectar una tuberculosis latente. El infliximab administra por vía intravenosa, de 3 a 5 mg/kg una vez, de nuevo 2 semanas más tarde y, a continuación, una vez al mes. La respuesta máxima puede demorar de 3 a 6 meses. El infliximab generalmente se combina con dosis bajas de metotrexato o azatioprina para prevenir la formación de anticuerpos contra él.
Puede considerarse la administración de adalimumab en pacientes con sarcoidosis ocular o cutánea que han sido tratados con éxito con infliximab pero que han desarrollado anticuerpos o reacciones a la infusión. El adalimumab se administra en dosis de 40 a 80 mg por vía subcutánea cada 1 a 2 semanas.
Otras consideraciones terapéuticas
En los pacientes que tienen bloqueos o arritmias ventriculares debido a la afectación cardíaca, se debe implantar un cardiodesfibrilador implantable y un marcapasos y deben recibir tratamiento con medicamentos.
Ningún fármaco disponible ha evitado de modo sistemático la fibrosis pulmonar.
El tratamiento de la hipertensión pulmonar asociada con sarcoidosis es sintomático con diuréticos y oxígeno suplementario. El papel de los vasodilatadores pulmonares en el tratamiento de la hipertensión pulmonar asociada con sarcoidosis no ha sido bien establecido; algunos estudios pequeños han sugerido su eficacia, pero se necesitan estudios de mayor envergadura para confirmarlo (1).
El trasplante de órganos es una opción para los pacientes con compromiso pulmonar, cardíaco o hepático en etapa terminal, aunque la enfermedad puede recidivar en el órgano trasplantado.
Los pacientes con sarcoidosis y deterioro moderado o grave de la función pulmonar o cardíaca tienen mayor riesgo de desarrollar formas graves y muerte por la infección por COVID-19. Al igual que en los pacientes con otras enfermedades pulmonares inflamatorias, durante la pandemia de COVID-19, los inmunosupresores deben usarse con precaución. Se recomienda con intensidad la adiministración de la vacuna contra SARS-CoV-2 en pacientes con sarcoidosis porque podría reducir la mortalidad y la gravedad de la enfermedad debido a la infección por COVID-19. Un ensayo clínico en curso tiene como objetivo evaluar mejor la eficacia de la vacunación contra el SARS-CoV-2 en pacientes con sarcoidosis y en los que toman inmunosupresores.
Referencia del tratamiento
1. Humbert M, Kovacs G, Hoeper MM, et al: 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 61(1): 1-144, 2023. doi:10.1183/13993003.00879-2022
Pronóstico de la sarcoidosis
Aunque la remisión espontánea es común, las manifestaciones y la gravedad de la enfermedad son muy variables, y muchos pacientes requieren corticosteroides para aliviar los síntomas o retardar el deterioro progresivo de la función orgánica en algún momento durante el curso de la enfermedad. Por lo tanto, es imperativo realizar el control seriado para poner de manifiesto la recidiva. Casi dos tercios de los pacientes con sarcoidosis finalmente logran la remisión con pocas o ninguna secuela. Alrededor del 50% de los pacientes presentan una remisión espontánea dentro de los primeros 3 años después del diagnóstico. Menos del 10% de estos pacientes recaen después de 2 años. Es probable que los pacientes que no presentan una remisión dentro de los 2-3 años sean propensos a tener la enfermedad crónica.
Se considera que la sarcoidosis es crónica en hasta un 30% de los pacientes y del 10 al 20% presentan secuelas permanentes. La enfermedad es mortal en el 1 a 5% de los pacientes, en general debida a una insuficiencia respiratoria causada por la fibrosis pulmonar y, con menor frecuencia, debida a una hemorragia pulmonar causada por aspergiloma. Sin embargo, en Japón, la miocardiopatía infiltrativa que causa arritmias e insuficiencia cardíaca es la causa más común de muerte.
El pronóstico es peor en los pacientes con sarcoidosis extrapulmonar y de raza negra. La remisión se produce en el 89% de los pacientes de raza blanca y el 76% de los de raza negra sin enfermedad extratorácica y en el 70% de los pacientes de raza blanca y el 46% de los de raza negra con enfermedad extratorácica.
Los signos de buen pronóstico son
Síndrome de Löfgren (tríada de poliartritis aguda, eritema nudoso y adenopatía hiliar)
Los signos de mal pronóstico son
Uveítis crónica
Lupus pernio
Hipercalcemia crónica
Neurosarcoidosis
Afectación cardíaca
Compromiso pulmonar extenso y/o desarrollo de hipertensión pulmonar
Hay poca diferencia demostrable a largo plazo entre los pacientes tratados y los no tratados; la recidiva es frecuente cuando finaliza el tratamiento.
Conceptos clave
La afectación sistémica y extrapulmonar son comunes con la sarcoidosis, pero > 90% de los pacientes adultos tienen afectación pulmonar.
Obtener un estudio de imagen del tórax, pero confirmar el diagnóstico mediante biopsia, por lo general aspiración transbronquial con aguja de un ganglio linfático mediastínico o hiliar, guiada por ecografía endobronquial.
Evaluar la gravedad pulmonar con las pruebas de función pulmonar y la oximetría de pulso en el ejercicio.
Estudios para el compromiso extrapulmonar con ECG, examen con lámpara de hendidura, pruebas de función renal y hepática, y determinación de calcio sérico y urinario.
Tratar a los pacientes con corticosteroides sistémicos cuando esté indicado (p. ej., síntomas graves, hipercalcemia, disminución progresiva de la función de los órganos, afectación cardíaca o neurológica).
Tratar con inmunosupresores si los pacientes no pueden tolerar las dosis moderadas de corticosteroides, la sarcoidosis es resistente a los mismos, o si se requieren corticosteroides a largo plazo.







