


La trombocitopenia inmunitaria es un trastorno hemorragíparo, en general sin anemia ni leucopenia. Por lo general, es crónica en adultos, pero suele ser aguda y autolimitada en niños. El tamaño del bazo es normal en ausencia de otra enfermedad subyacente. El diagnóstico suele ser clínico y se basa en la exclusión de otras causas de trombocitopenia (p. ej., infecciones por HIV y hepatitis C). El tratamiento consiste en corticosteroides, esplenectomía, inmunosupresores, fármacos agonistas de los receptores de trombopoyetina o el inhibidor esplénico de la tirosina cinasa fostamatinib. En hemorragias potencialmente fatales, pueden utilizarse transfusiones de plaquetas, corticosteroides IV, inmunoglobulina anti-D IV o inmunoglobulina IV en forma individual o combinada.
(Véase también Generalidades sobre los trastornos plaquetarios).
Por lo general, la trombocitopenia inmunitaria se debe al desarrollo de un autoanticuerpo dirigido contra un antígeno plaquetario estructural. Estos anticuerpos antiagregantes plaquetarios aumentan la destrucción de las plaquetas, por lo general en el bazo, e inhiben la producción y la liberación de plaquetas por los megacariocitos. En la trombocitopenia inmunitaria de la infancia, la formación del autoanticuerpo puede ser desencadenada por los antígenos virales. El desencadenante en adultos es desconocido, aunque en algunos países (p. ej., Japón, Italia), se ha asociado a la trombocitopenia inmunitaria con la infección por Helicobacter pylori, y el tratamiento de la infección logró la remisión de la trombocitopenia inmunitaria (1). La infección por COVID-19 rara vez causa PTI, pero la vacunación contra la COVID-19 puede empeorar la trombocitopenia en el 2 al 12% de los pacientes con PTI. La trombocitopenia inmunitaria tiende a empeorar durante el embarazo y aumenta el riesgo de morbilidad materna.
Referencia general
1. Kuter DJ: The treatment of immune thrombocytopenia (ITP)—focus on thrombopoietin receptor agonists. Annals of Blood Volume 6 March 2021
Signos y síntomas de la trombocitopenia inmunitaria
Aunque a menudo son asintomáticos y se identifican solo por un recuento bajo de plaquetas en un ensayo de rutina, cuando se producen síntomas y signos de trombocitopenia inmunitaria, estos son
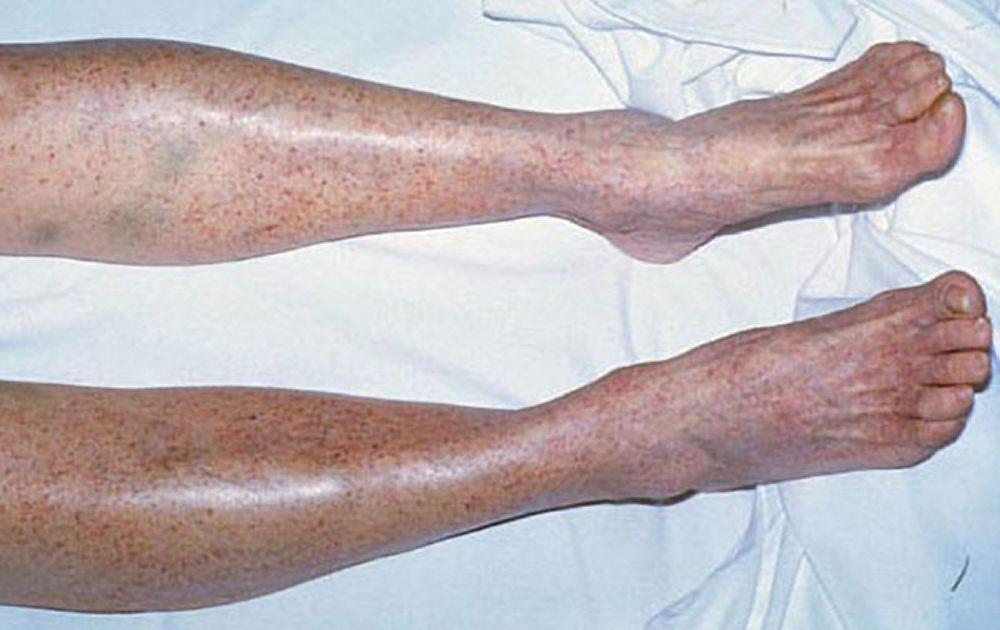
Petequias
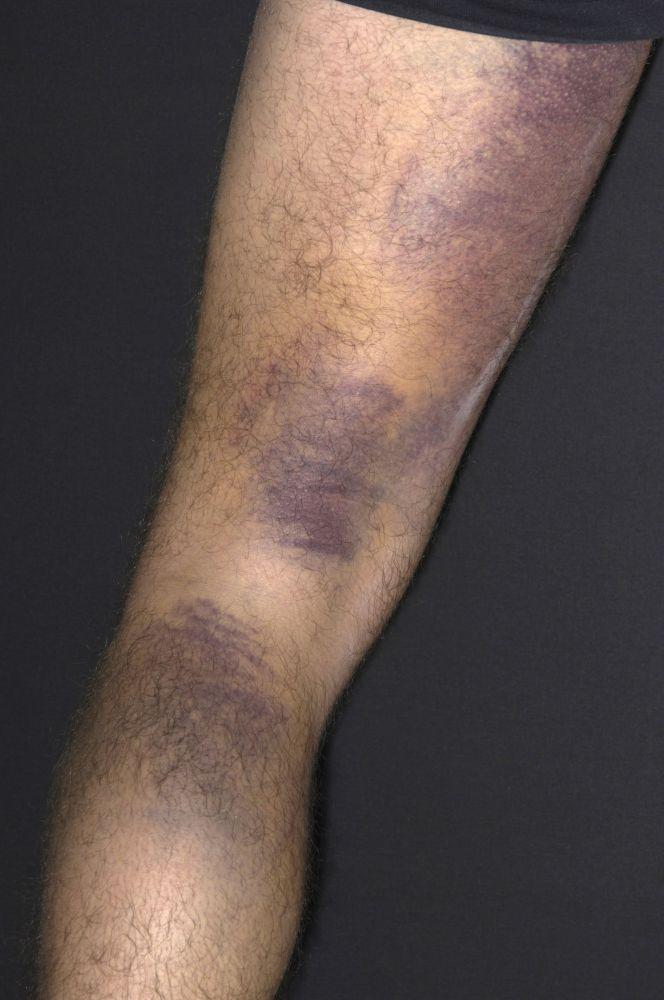
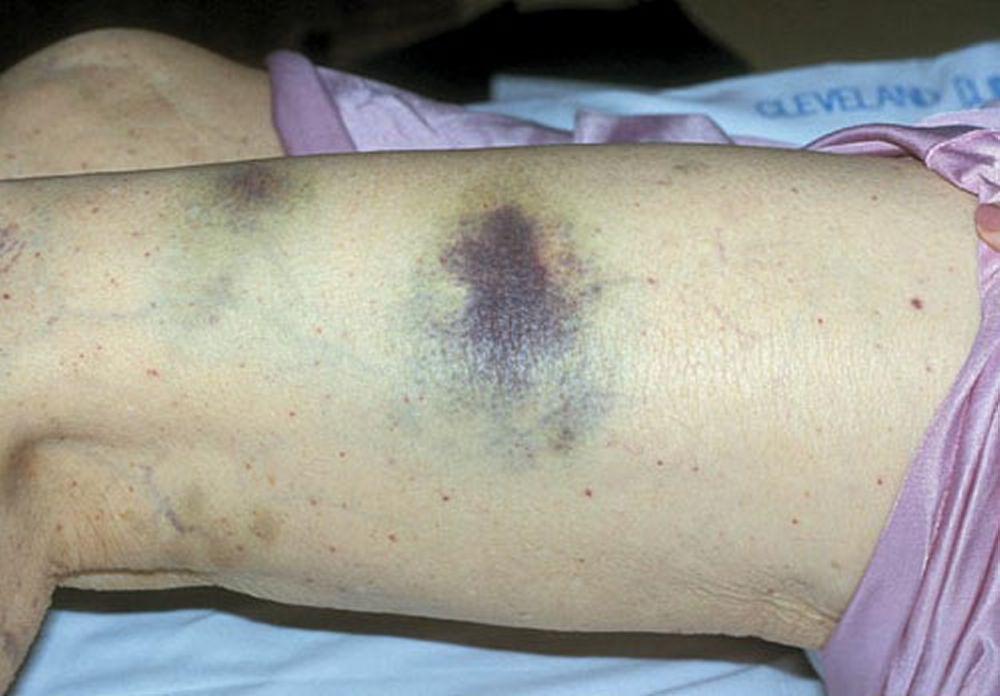
Púrpura y/o equimosis
Sangrado de la mucosa
Aumento del sangrado menstrual
La hemorragia digestiva y la hematuria macroscópicas son menos frecuentes. El bazo es de tamaño normal, a menos que esté agrandado por una infección viral o una anemia hemolítica autoinmune coexistente (síndrome de Evans). Al igual que los otros trastornos de aumento de la destrucción de las plaquetas, la trombocitopenia inmunitaria también se asocia con un mayor riesgo de trombosis.
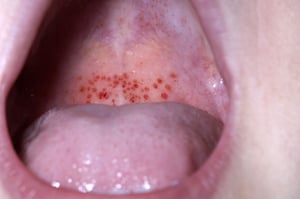
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
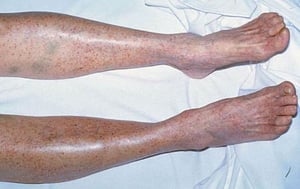
Con autorización del editor. From Deitcher S. En Atlas of Clinical Hematology. Editado por JO Armitage. Philadelphia, Current Medicine, 2004.
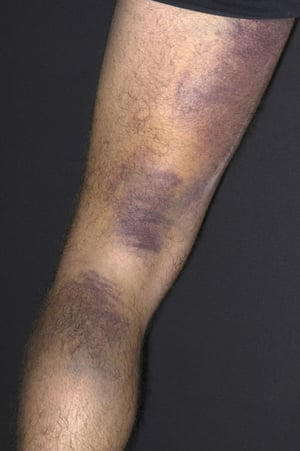
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
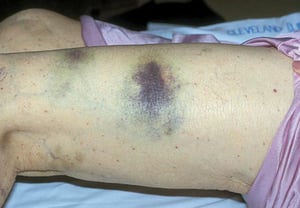
Con autorización del editor. De Deitcher S. En Atlas of Clinical Hematology. Editado por JO Armitage. Philadelphia, Current Medicine, 2004.

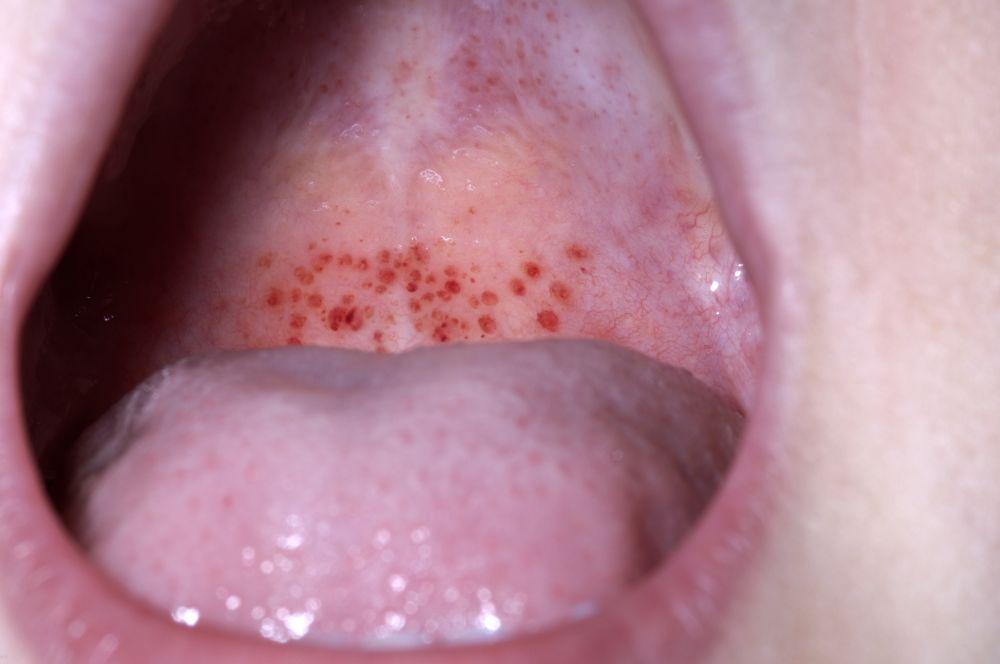
DR P. MARAZZI/SCIENCE PHOTO LIBRARY

Con autorización del editor. From Deitcher S. En Atlas of Clinical Hematology. Editado por JO Armitage. Philadelphia, Current Medicine, 2004.

DR P. MARAZZI/SCIENCE PHOTO LIBRARY

Con autorización del editor. De Deitcher S. En Atlas of Clinical Hematology. Editado por JO Armitage. Philadelphia, Current Medicine, 2004.
Diagnóstico de la trombocitopenia inmunitaria
Hemograma con recuento de plaquetas y frotis de sangre periférica
Pocas veces, aspiración de médula ósea
Exclusión de otros trastornos trombocitopénicos
Se sospecha trombocitopenia inmunitaria en pacientes con trombocitopenia aislada (es decir, hemograma completo y frotis de sangre periférica por lo demás normal). Como las manifestaciones de tromboticopenia inmunitaria son inespecíficas, deben descartarse otras causas de trombocitopenia aislada reversibles (p. ej., fármacos, alcohol, trastornos linfoproliferativos, otras enfermedades autoinmunitarias, infecciones virales) por evaluación clínica y estudios complementarios apropiados. Por lo general, los pacientes tienen estudios de coagulación, pruebas hepáticas y pruebas para la infección por el virus de la hepatitis C y el HIV. El frotis de sangre periférica debe ser revisado para evaluar el tamaño de las plaquetas y su granularidad y ayudar a excluir otras causas importantes de trombocitopenia, como púrpura trombocitopénica trombótica (PTT), trombocitopenia hereditaria y leucemia. Las pruebas para anticuerpos antiplaquetarios pueden ayudar al diagnóstico en algunos casos (1). La fracción de plaquetas inmaduras puede estar elevada en la trombocitopenia inmunitaria cuando el recuento de plaquetas es < 50.000/mcL (< 50 × 109/L).
No se requiere examen de médula ósea para arribar al diagnóstico, pero se practica si el hemograma o el frotis revelan otras alteraciones además de la trombocitopenia, cuando las manifestaciones clínicas no son típicas o si los pacientes no responden a los tratamientos estándares (p. ej., corticosteroides). En la PTI, el examen de médula ósea revela números normales o posiblemente aumentados de megacariocitos en una muestra de médula ósea por lo demás normal.
Referencia del diagnóstico
1. Al-Samkari H, Rosovsky RP, Karp Leaf RS: A modern reassessment of glycoprotein-specific direct platelet autoantibody testing in immune thrombocytopenia. Blood Adv 4(1):9–18, 2020. doi: 10.1182/bloodadvances.2019000868
Pronóstico de la trombocitopenia inmunitaria
Por lo general, los niños se recuperan espontáneamente, aun de trombocitopenias graves, en varias semanas o meses.
En adultos, la remisión espontánea se produce en menos del 10%. Al finalizar el tratamiento inicial, alrededor de un tercio de los pacientes experimenta remisión. Hasta el 75% de los pacientes mejora dentro de los 5 años (1). Sin embargo, muchos pacientes presentan enfermedad leve y estable (es decir, recuentos de plaquetas > 30.000/mcL [> 30 × 109/L]) con un sangrado mínimo o nulo; con frecuencia se descubren en el recuento automatizado de plaquetas que, en la actualidad, se realiza rutinariamente con el hemograma completo.
Referencia del pronóstico
1. Sailer T, Lechner K, Panzer S, et al: The course of severe autoimmune thrombocytopenia in patients not undergoing splenectomy. Haematologica 91:1041–1045, 2006.
Tratamiento de la trombocitopenia inmunitaria
Corticosteroides orales
Inmunoglobulina IV (IVIG)
Inmunoglobulina anti-D IV
En ocasiones, esplenectomía
Agonistas del receptor de trombopoyetina (TPO-RA)
Rituximab
Fostamatinib
Otros inmunosupresores
En la hemorragia grave, IVIG, inmunoglobulina anti-D IV, corticosteroides IV y/o transfusiones de plaquetas
Las pautas para el año 2019 ya están disponibles (1, 2). Los pacientes asintomáticos con un recuento de plaquetas > 30.000/mcL (> 30 × 109/L) y sin sangrado no requieren tratamiento y pueden controlarse con conducta expectante.
Por lo general, los adultos con diagnóstico reciente de tromboticopenia inmunitaria con hemorragia y un recuento de plaquetas < 30.000/mcL (< 30 × 109/L) reciben inicialmente un corticoide oral (p. ej., prednisona 1 mg/kg por vía oral 1 vez al día). Un régimen alternativo de corticosteroides, probablemente igual de eficaz, consiste en dexametasona 40 mg por vía oral 1 vez al día durante 4 días. En la mayoría de los pacientes, el recuento de plaquetas aumenta en 2 a 5 días. Sin embargo, en algunos pacientes, la respuesta puede tardar de 2 a 4 semanas. Cuando se reduce la dosis del corticosteroide después de la respuesta, la mayoría de los pacientes adultos recaen. Los tratamientos repetidos con corticosteroides pueden ser eficaces, pero aumentan el riesgo de efectos adversos. Los corticosteroides no suelen continuarse más allá de los primeros meses; pueden probarse otros fármacos en un intento de evitar la esplenectomía. Si el tratamiento médico es eficaz, la mayoría de las guías recomienda continuarlo durante al menos un año antes de considerar la esplenectomía.
Asimismo, pueden administrarse corticosteroides orales, IGIV o inmunoglobulina anti-D IV cuando se requiere un aumento transitorio del recuento plaquetario para extracciones dentarias, parto, cirugía u otros procedimientos invasivos. Los agonistas de los receptores de trombopoyetina (TPO-RA, p. ej., romiplostim, eltrombopag, avatrombopag) también pueden usarse antes de los procedimientos invasivos, pero no deben usarse para el parto. La IVIG y la inmunoglobulina anti-D IV también son útiles para la hemorragia potencialmente letal en la tromboticopenia inmunitaria, pero rara vez se usa para el tratamiento crónico porque su respuesta puede durar solo unos días o semanas.
La esplenectomía puede lograr una remisión completa en aproximadamente dos tercios de los pacientes que recaen después de la terapia con corticosteroides inicial. La esplenectomía generalmente se reserva para pacientes con trombocitopenia grave (p. ej., < 15.000/mcL [< 15 × 109/L]) en los que el riesgo de sangrado no se puede controlar con terapia médica o en aquellos cuya enfermedad persiste después de 12 meses. Si la trombocitopenia se puede controlar con terapias farmacológicas de segunda línea, la esplenectomía suele no ser necesaria (1, 2). La esplenectomía produce un aumento del riesgo de trombosis e infección (en particular por bacterias encapsuladas como neumococo); los pacientes requieren vacunación contra Streptococcus pneumoniae, Haemophilus influenzae, y Neisseria meningitidis (idealmente > 2 semanas antes del procedimiento).
Terapias médicas de segunda línea
Hay terapias farmacológicas de segunda línea están disponibles para los pacientes con trombocitopenia inmunitaria
Que intentan postergar la esplenectomía con la esperanza de una remisión espontánea?
¿Quiénes no son candidatos o rechazan la esplenectomía?
En quien la esplenectomía no ha sido efectiva
Estos pacientes suelen tener recuentos de plaquetas < 10.000 a 20.000/mcL (< 10 a 20 × 109/L), (y por lo tanto están en riesgo de sangrado). Las terapias farmacológicas de segunda línea incluyen agonistas del receptor de trombopoyetina (TPO-RAs), rituximab, fostamatinib u otros fármacos inmunosupresores.
Los agonistas del receptor de la trombopoyetina, como romiplostim, en dosis de 1 a 10 mcg/kg por vía subcutánea 1 vez por semana, el eltrombopag, en dosis de 25 a 75 mg por vía oral 1 vez al día y el avatrombopag 20 mg/día por vía oral tienen tasas de respuesta > 85%. Los agonistas del receptor de trombopoyetina a menudo necesitan ser administrados continuamente para mantener un recuento de plaquetas > 50.000/mcL (> 50 × 109/L), pero datos sugieren que un tercio de los adultos logrará una remisión libre de tratamiento después de 1 año y > 50% después de 2 años.
El rituximab (375 mg/m2 IV 1 vez/semana durante 4 semanas) tiene una tasa de respuesta del 57%, pero sólo el 21% de los pacientes adultos permanecen en remisión después de 5 años (3). Los esquemas de dosificación alternativos también son eficaces (p. ej., dosis de 1.000 mg IV administradas con 2 semanas de diferencia). El rituximab puede afectar la capacidad de respuesta a la vacunación durante 6 a 12 meses.
El fostamatinib, un inhibidor de la tirosina cinasa del bazo, se asocia con una tasa de respuesta del 18%. La dosis es de 100 mg por vía oral 2 veces al día y aumenta a 150 mg después de 1 mes si el recuento de plaquetas no ha aumentado a > 50.000/mcL (> 50 × 109/L—4).
Puede requerirse inmunosupresión más intensiva con fármacos como ciclofosfamida, ciclosporina, micofenolato y azatioprina en pacientes con trombocitopenia grave, sintomática, que no responden a otros fármacos.
Hemorragia que pone en peligro la vida en la trombocitopenia inmunitaria
En niños o adultos con trombocitopenia inmunitaria y hemorragia potencialmente letal, se intenta un bloqueo fagocítico rápido administrando IGIV en dosis de 1 g/kg 1 vez al día durante 1-2 días o, en pacientes Rh positivos, una dosis única de inmunoglobulina anti-D 75 mcg/kg. La inmunoglobulina anti-D IV intravenosa solo es eficaz en pacientes que no se han sometido a una esplenectomía y puede asociarse con complicaciones graves, como hemólisis grave y coagulación intravascular diseminada. Por lo general, este tratamiento induce un aumento del recuento de plaquetas en el término de 2 a 4 días, pero el recuento permanece alto sólo durante 2-4 semanas.
La metilprednisolona en alta dosis (1 g IV 1 vez al día durante 3 días) es más fácil de administrar que la IGIV o la inmunoglobulina anti-D IV pero puede ser menos eficaz. Los pacientes con trombocitopenia inmunitaria y hemorragia potencialmente fatal también reciben transfusiones de plaquetas. Éstas no se utilizan en forma profiláctica.
La vincristina (1,4 mg/m2, dosis máxima de 2 mg) también se ha utilizado en situaciones de emergencia pero puede producir neuropatía con la administración repetida.
El uso temprano de TPO-RA también puede ser eficaz en combinación con las terapias anteriores (5, 6).
Tratamiento de niños con trombocitopenia inmunitaria
Por lo general, el tratamiento de los niños con trombocitopenia inmunitaria es sintomático porque la mayoría se recupera en forma espontánea. Aun después de meses o años de trombocitopenia, la mayoría de los niños presentan remisiones espontáneas. Si sobreviene una hemorragia mucosa, pueden administrarse corticosteroides o IGIV. El uso de corticosteroides e IGIV es controvertido, porque el aumento del recuento de plaquetas puede no mejorar la evolución clínica. Rara vez se realiza una esplenectomía en niños. Sin embargo, si la trombocitopenia es grave y sintomática durante > 6 meses, se debe considerar agonistas del receptor de trombopoyetina (p. ej., romiplostim, eltrombopag).
Referencias del tratamiento
1. Neunert C, Terrell DR, Arnold DM, et al: American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv 3(23):3829–3866, 2019. doi: 10.1182/bloodadvances.2019000966
2. Provan D, Arnold DM, Bussel JB, et al: Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv 3(22):3780–3817, 2019. doi: 10.1182/bloodadvances.2019000812
3. Patel VL, Mahevas M, Lee SY, et al: Outcomes 5 years after response to rituximab therapy in children and adults with immune thrombocytopenia. Blood 119:5989–5995, 2012. doi: 10.1182/blood-2011-11-393975
4. Bussel J, Arnold DM, Grossbard E, et al: Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo‐controlled trials. Am J Hematol 93: 921–930, 2018. doi: 10.1002/ajh.25125
5. Kuter DJ, Tarantino MD, Lawrence T: Clinical overview and practical considerations for optimizing romiplostim therapy in patients with immune thrombocytopenia. Blood Rev 49:100811, 2021. doi: 10.1016/j.blre.2021.100811
6. Lozano ML, Godeau B, Grainger J, et al: Romiplostim in adults with newly diagnosed or persistent immune thrombocytopenia. Expert Rev Hematol 13(12):1319–1332, 2020. doi: 10.1080/17474086.2020.1850253
Conceptos clave
En la tromboticopenia inmunitaria, el sistema inmunitario destruye las plaquetas en la circulación y al mismo tiempo y puede atacar a los megacariocitos de la médula ósea, reduciendo de ese modo la producción de plaquetas.
Se deben descartar otras causas de trombocitopenia aislada (p. ej., los fármacos, el alcohol, los trastornos linfoproliferativos, otras enfermedades autoinmunes, infecciones virales).
Los niños por lo general tienen una remisión espontánea; en los adultos, la remisión espontánea puede ocurrir durante el primer año, pero es menos común (alrededor del 33%) que en los niños.
Los corticosteroides (y a veces inmunoglobulina intravenos [IGIV] o inmunoglobulina anti-D IV) son tratamientos de primera línea para el sangrado o la trombocitopenia grave.
La biopsia de médula ósea generalmente no es necesaria a menos que se identifiquen otras anomalías preocupantes en los glóbulos rojos o blancos o en candidatos a la esplenectomía que no han respondido al tratamiento estándar con corticosteroides o IVIG.
Los agonistas de los receptores de trombopoyetina son muy eficaces para mantener un recuento plaquetario seguro en > 85% de los adultos.
La infección por COVID-19 rara vez causa PTI, pero la vacunación contra la COVID-19 puede empeorar la trombocitopenia en el 2 al 12% de los pacientes con PTI.
La esplenectomía es a menudo eficaz, pero se reserva para pacientes en los que el tratamiento médico es ineficaz o aquellos cuya enfermedad persiste después de 12 meses.
Se administra transfusión de plaquetas solo para la hemorragia potencialmente fatal.









