


La osteogénesis imperfecta es un trastorno hereditario del colágeno que causa fragilidad ósea anormal difusa y, en ocasiones, se acompaña de hipoacusia neurosensorial, escleróticas azules, dentinogénesis imperfecta e hipermovilidad articular. El diagnóstico suele ser clínico. El tratamiento incluye hormona de crecimiento para algunos tipos, bifosfonatos y denosumab.
Existen 4 tipos principales de osteogénesis imperfecta (1):
I: Escleróticas azules leves
II: neonatal letal, escleróticas azules
III: progresivo, color variable de las escleróticas
IV: Variable y deformante, color normal de las escleróticas
Su modo de herencia suele ser autosómica dominante. El 90% de las personas que tienen uno de los principales tipos tienen mutaciones en los genes que codifican las cadenas pro-alfa del procolágeno tipo I (un componente estructural de los huesos, los ligamentos y los tendones), COL1A1 o COL1A2.
Hay una serie de otros tipos más raros (tipos V a XXI), que son causados por mutaciones de diferentes genes.
Referencia
1. Van Dijk FS, Sillence DO: Osteogenesis imperfecta: Clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A 164A(6):1470–1481, 2014. doi: 10.1002/ajmg.a.36545. Clarification and additional information. Am J Med Genet A 167A(5):1178, 2015. doi: 10.1002/ajmg.a.36784
Signos y síntomas de la psteogénesis imperfecta
El 50-65% de los pacientes con osteogénesis imperfecta presenta hipoacusia, que puede aparecer en cualquiera de los 4 tipos principales..
El tipo I es el más leve. En algunos pacientes, los signos y síntomas se limitan a escleróticas azules (debido a una deficiencia del tejido conectivo que permite la visualización a través de éste de los vasos subyacentes) y dolor musculoesquelético por hipermovilidad articular. En la infancia, son posibles las fracturas recurrentes.
El tipo II (tipo neonatal letal u osteogénesis imperfecta congénita) es la forma más grave y es letal. Múltiples fracturas congénitas determinan acortamiento de los miembros. Las escleróticas son azules. El cráneo es blando y, al palparlo, produce la sensación de una bolsa de huesos. Como el cráneo es blando, el traumatismo durante el parto puede provocar hemorragia intracraneal y muerte fetal, o los recién nacidos pueden presentar muerte súbita durante los primeros días o semanas de vida.

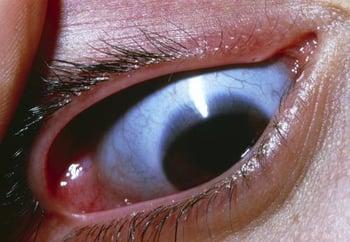
El tipo III es progresivo y la forma no letal más grave de osteogénesis imperfecta. Los pacientes con tipo III tienen talla baja, curvatura de la columna y múltiples fracturas recurrentes. La macrocefalia con cara triangular y deformidades torácicas son frecuentes. El color de las escleróticas es variable.


El tipo IV es de gravedad intermedia. La tasa de supervivencia es alta. Este tipo es variable y deformante. Los huesos se fracturan fácilmente en la infancia antes de la adolescencia. Por lo general, las escleróticas son de color normal. La talla es moderada-baja. El diagnóstico exacto es importante porque estos pacientes pueden beneficarse con el tratamiento.
Diagnóstico de la osteogénesis imperfecta
Evaluación clínica
A veces análisis del procolágeno tipo I o pruebas genéticas
Por lo general, el diagnóstico de la osteogénesis imperfecta es clínico, pero no hay ningún criterio estandarizado.
Cuando el diagnóstico es dudoso, puede recurrirse al análisis del procolágeno tipo I de fibroblastos cultivados (obtenidos por biopsia de piel) o al análisis de la secuencia de los genes de COL1A1 y COL1A2.
La ecografía de nivel II permite la detección intrauterina de osteogénesis imperfecta grave.
Tratamiento de la osteogénesis imperfecta
Hormona de crecimiento
Bisfosfonatos
A veces denosumab
A veces, vitamina D
La hormona de crecimiento ayuda a los niños que responden al crecimiento (tipos I y IV).
El tratamiento con bisfosfonatos tiene como objetivo aumentar la densidad ósea y disminuir el dolor óseo y el riesgo de fractura (1). Se usa pamidronato intravenoso (0,5 a 3 mg/kg 1 vez al día durante 3 días, repetido si es necesario cada 4 a 6 meses) o alendronato oral (1 mg/kg, 20 mg máximo, 1 vez al día).
El denosumab es un potente inhibidor de la resorción ósea osteoclástica y suele administrarse por vía inyectable. Los estudios han mostrado que este medicamento es beneficioso en algunos pacientes con osteogénesis imperfecta (2).
Se debe administrar un suplemento de vitamina D a las personas con deficiencia de esta hormona.
La cirugía ortopédica, la fisioterapia y la terapia ocupacional ayudan a prevenir fracturas y mejorar la función.
En algunos casos de hipoacusia, está indicado el implante coclear.
Referencias del tratamiento
1. Dwan K, Phillipi CA, Steiner RD, Basel D: Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev CD005088, 2016. doi: 10.1002/14651858.CD005088.pub4
2. Li G, Jin Y, Levine MAH, et al: Systematic review of the effect of denosumab on children with osteogenesis imperfecta showed inconsistent findings. Acta Paediatr 107(3):534–537, 2018. doi: 10.1111/apa.14154
Más información
El siguiente recurso en inglés puede ser útil. Tenga en cuenta que el MANUAL no es responsable por el contenido de este recurso.
Osteogenesis Imperfecta (OI) Foundation: An organization providing support, education, and research information about OI









