







La chorée de Huntington est une maladie héréditaire qui débute par des saccades ou des spasmes involontaires occasionnels ; elle évolue vers la survenue de mouvements involontaires (chorée et athétose) de plus en plus marqués associés à une détérioration mentale et le décès.
Dans la chorée de Huntington, les parties du cerveau qui permettent de coordonner et d’harmoniser les mouvements se dégradent.
Les mouvements deviennent saccadés et non coordonnés, et la fonction cognitive, y compris la maîtrise de soi et la mémoire, se détériore.
Le diagnostic s’appuie sur les symptômes, les antécédents familiaux, l’imagerie du cerveau et une analyse génétique.
Des médicaments peuvent soulager certains des symptômes, mais la maladie est évolutive, conduisant finalement au décès.
(Voir également Présentation des troubles du mouvement.)
La chorée de Huntington affecte 1 à 10 personnes sur 100 000. Le nombre de personnes touchées varie selon la région du monde où elles vivent. Elle touche les deux sexes de manière égale.
Le gène responsable de cette maladie est dominant. C’est-à-dire qu’une seule copie du gène anormal, hérité de l’un des parents, suffit donc à provoquer la maladie. Par conséquent, chez les enfants d’une personne atteinte de la chorée de Huntington, le risque de développer la maladie est de 50 %.
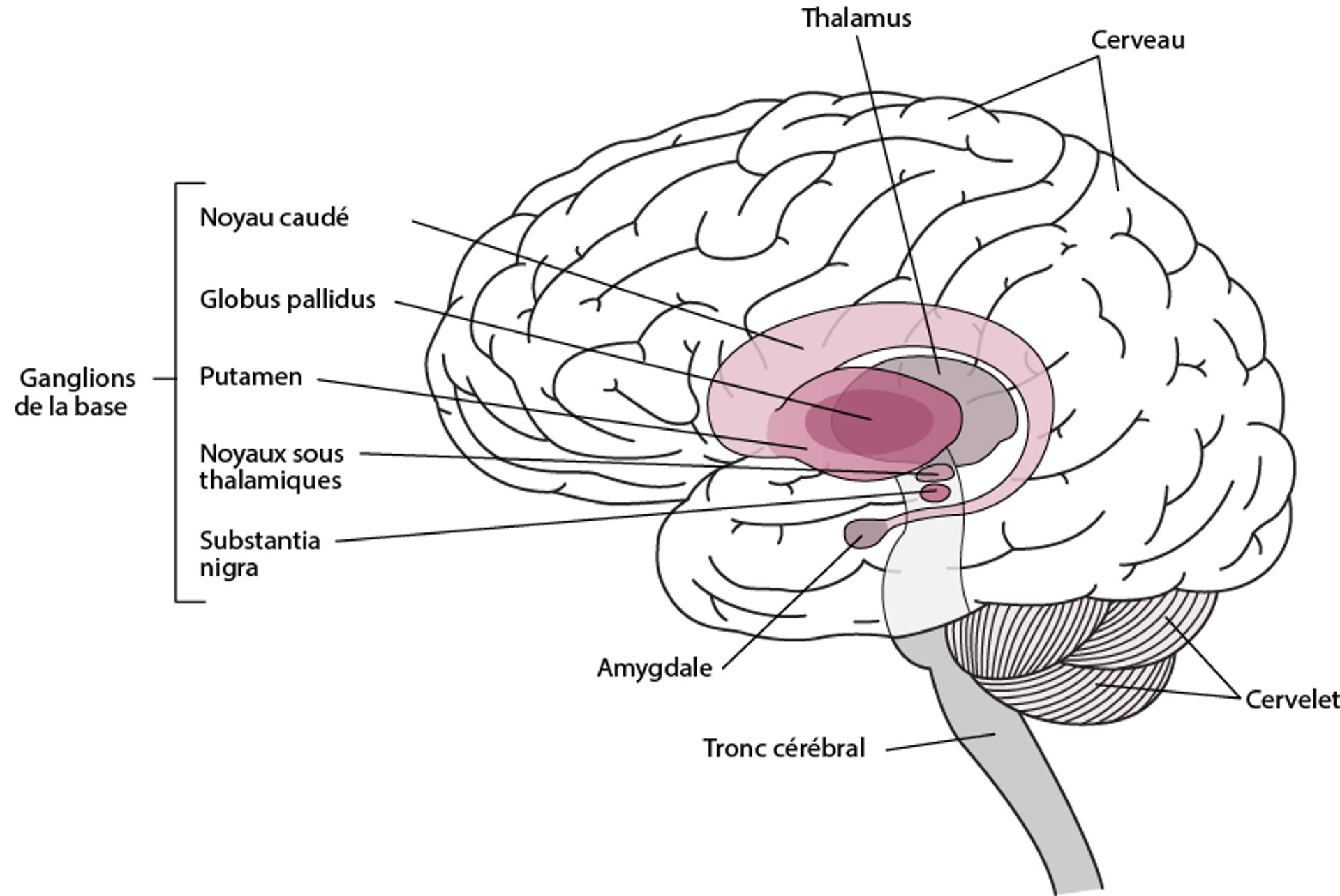
La maladie de Huntington est causée par une dégénérescence progressive de certaines parties des noyaux basaux, appelées noyau caudé et putamen. Les noyaux basaux sont des groupes des cellules nerveuses situées en profondeur, à la base du cerveau. Ils coordonnent et harmonisent les mouvements.
Localisation des noyaux basaux
Les noyaux basaux sont des groupes de cellules nerveuses situés dans les profondeurs du cerveau. À savoir :
Les noyaux basaux permettent d’initier et d’harmoniser les mouvements musculaires, de supprimer les mouvements involontaires et de coordonner les changements de posture. |

Symptômes de la chorée de Huntington
Le plus souvent, les symptômes de la chorée de Huntington se développent de façon insidieuse, débutant généralement entre l’âge de 35 et 40 ans, mais parfois avant l’âge adulte.
Au début de la chorée de Huntington, le visage, le tronc et les membres bougent de manière involontaire et brusque. La personne peut d’abord masquer les mouvements anormaux en les transformant en mouvements volontaires, de sorte que les mouvements anormaux sont à peine perceptibles. Cependant, avec le temps, les mouvements deviennent plus visibles.
Les muscles peuvent se contracter brièvement et rapidement, ce qui entraîne une saccade des bras ou d’autres parties du corps, parfois plusieurs fois d’affilée.
Les personnes peuvent marcher d’une manière cadencée ou exagérément désinvolte, comme une marionnette. Elles grimacent, bougent de façon irrégulière les membres et clignent des yeux. Les mouvements sont irréguliers et ralentis. À un stade avancé, l’ensemble du corps est atteint, rendant extrêmement difficile le fait de marcher, de rester assis(s), de s’alimenter, de parler, de déglutir et de s’habiller.
Des changements mentaux se produisent fréquemment avant ou pendant le développement des mouvements anormaux. Ces modifications sont insidieuses au début. Les personnes peuvent devenir progressivement irritables, excitables et agitées. Elles peuvent perdre tout intérêt pour les activités de la vie quotidienne. Les personnes ne parviennent pas à contrôler leurs réactions émotionnelles, elles perdent leur sang-froid, sont irritées et parfois désespérées.
Lorsque la chorée de Huntington évolue, les personnes se comportent de manière irresponsable et errent souvent sans but. Avec les années, elles perdent la mémoire et leurs capacités de jugement. Elles peuvent devenir très déprimées et faire des tentatives de suicide. Elles peuvent également devenir anxieuses ou développer un trouble obsessionnel compulsif.
Dans les stades évolués de la maladie, on observe une démence prononcée, et l’alitement est nécessaire. Une aide à plein temps au domicile ou un placement en maison de santé est alors nécessaire. Le décès survient habituellement 13 à 15 ans après le début des symptômes.
Diagnostic de la chorée de Huntington
Examen clinique, confirmé par des analyses génétiques
Tomodensitométrie ou imagerie par résonance magnétique
Le diagnostic de la maladie de Huntington peut être difficile au début, car les symptômes sont insidieux. On peut suspecter cette affection en se basant sur les symptômes et les antécédents familiaux. Les médecins doivent être informés sur les membres de la famille qui ont eu des troubles psychiatriques ou pour lesquels un diagnostic de maladie neurologique (comme la maladie de Parkinson) ou de maladie psychiatrique (comme la schizophrénie) a été posé, car ils étaient peut-être atteints de la maladie de Huntington sans qu’elle soit diagnostiquée.
On procède à une tomodensitométrie (TDM) ou une imagerie par résonance magnétique (IRM) afin de déceler une éventuelle dégénérescence des noyaux basaux et d’autres parties du cerveau généralement touchées par la maladie, mais également afin d’éliminer d’autres maladies.
On effectue une analyse génétique pour confirmer le diagnostic. L’analyse génétique et des conseils sont importants pour les personnes qui ont des antécédents familiaux de la maladie, mais qui ne présentent pas de symptômes, car ces personnes sont susceptibles d’avoir des enfants avant que les symptômes apparaissent. Ces personnes doivent être soumises au conseil génétique avant de réaliser cette analyse génétique. Elles sont orientées vers les centres qui sont spécialisés dans les problèmes éthiques et psychologiques complexes concernés.
Traitement de la chorée de Huntington
Des antipsychotiques et d’autres médicaments pour soulager les symptômes.
Dès que possible après le diagnostic, les personnes souffrant de la chorée de Huntington doivent établir des directives préalables de prise en charge pour la phase terminale.
La chorée de Huntington est incurable. Néanmoins, certains médicaments, notamment les antipsychotiques (tels que la chlorpromazine, l’halopéridol, la rispéridone et l’olanzapine) peuvent permettre de contrôler l’agitation. Les médicaments qui réduisent la quantité de dopamine (comme la tétrabénazine, la deutétrabenazine et l’antihypertenseur réserpine) peuvent permettre d’arrêter (supprimer) les mouvements anormaux.
Des antidépresseurs peuvent être utilisés pour traiter la dépression, le cas échéant.
Les médecins proposent une consultation en génétique et des analyses génétiques aux personnes ayant des parents ou des frères et sœurs atteints de chorée de Huntington. Une consultation génétique doit être proposée avant l’analyse génétique, car les conséquences de la chorée de Huntington sont très graves. Cette consultation est particulièrement importante pour les femmes en âge de procréer et les hommes qui envisagent de devenir pères.
Informations supplémentaires
Il s’agit d’une ressource en anglais qui peut être utile. Veuillez noter que le MANUEL n’est pas responsable du contenu de ces ressources.
Genetics Home Reference: Chorée de Huntington : Ce site Internet décrit la chorée de Huntington et explique ses causes et la manière dont elle est transmise. Il fournit également des liens vers son diagnostic et son traitement.




