







Le duodénum peut être obstrué par atrésie, par sténose et par une compression exercée par une masse extrinsèque.
(Voir aussi Revue générale des anomalies gastro-intestinales congénitales.)
Atrésie duodénale
L'atrésie duodénale est la 2e localisation par ordre de fréquence des atrésies du tube digestif. La fréquence est de 1 sur 10 000 naissances vivantes (1). L'atrésie duodénale est due à un défaut de canalisation du duodénum embryonnaire. Ce défaut peut être lié à un événement ischémique ou à des facteurs génétiques.
L'atrésie duodénale, contrairement à d'autres atrésies intestinales, est souvent associée à d'autres anomalies congénitales; environ 25 à 40% des nourrissons qui ont une atrésie duodénale ont un syndrome de Down (1). D'autres anomalies associées comprennent le syndrome VACTERL (anomalies vertébrales, atrésie anale, malformations cardiaques, fistuletrachéo-œsophagienne, anomalies rénales, aplasie radiale et anomalies des membres), la malrotation, le pancréas annulaire, les anomalies des voies biliaires et les anomalies mandibulo-faciales.
Le diagnostic d'atrésie duodénale est suspecté avant la naissance en cas d'hydramnios et/ou de dilatation de l'estomac. L'échographie prénatale peut détecter un signe de bulle double (une grande bulle gastrique et une plus petite bulle duodénale proximale) dans jusqu'à 80% des cas (2).

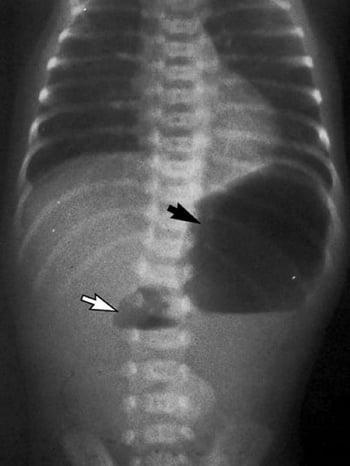
Après la naissance, les nourrissons atteints d'atrésie duodénale présentent initialement des difficultés à s'alimenter et des vomissements souvent bilieux. Le diagnostic est suspecté sur la clinique et la présence classique d'une image rx en "double bulle", une bulle gastrique et l'autre dans le duodénum proximal; le reste du tube digestif est peu, voire pas, aéré. Bien qu'une opacification gastro-intestinale haute permette d'établir un diagnostic de certitude, cet examen doit être réalisé avec prudence par un radiologue ayant l'expérience de cette procédure chez l'enfant, afin d'éviter toute fausse route, et il n'est pas habituellement nécessaire si une chirurgie doit avoir lieu immédiatement. Si la chirurgie doit être retardée (p. ex., par d'autres problèmes médicaux, comme un syndrome de détresse respiratoire, ou la nécessité de stabiliser le nourrisson), un lavement baryté doit être effectué pour confirmer que le signe de la double bulle n'est pas dû à une malrotation.
Une fois la maladie suspectée, les nourrissons doivent être laissés à jeun et une sonde nasogastrique doit être placée afin de décomprimer l'estomac.
Le traitement curateur est chirurgical.
Sténose duodénale
Cette anomalie est moins fréquente que l'atrésie duodénale mais se manifeste de façon similaire et nécessite une intervention chirurgicale. De même que l'atrésie, la sténose duodénale est souvent associée au syndrome de Down.
Kyste du cholédoque
Un kyste cholédocien peut entraîner une occlusion duodénale par compression extrinsèque. L'incidence varie d'environ 1 personne sur 100 000 aux États-Unis ou en Europe à 1 personne sur 13 000 au Japon (3). Plus de la moitié des cas signalés se produisent au Japon. Certaines données probantes sont en faveur d'une augmentation de l'incidence des kystes cholédociens.
Les nourrissons porteurs d'un kyste du cholédoque présentent généralement initialement la triade douleur abdominale (un signe très difficile à identifier chez le nouveau-né), une masse de l'hypochondre droit et un ictère. Si le kyste est volumineux, il peut également entraîner différents degrés d'occlusion duodénale. Les nouveau-nés peuvent présenter une cholestase. Dans certains cas, une pancréatite est un signe associé.
Les kystes du cholédoque sont le plus souvent identifiés par l'échographie. Ces kystes peuvent être définis plus avant en utilisant la cholangiopancréatographie par résonance magnétique, la CPRE (cholangiopancréatographie rétrograde endoscopique) ou l'écho-endoscopie.
Le traitement du kyste cholédocien est chirurgical et nécessite une exérèse complète du kyste du fait du risque élevé (20 à 30%) de développer un cancer dans le reliquat du kyste (4). La procédure chirurgicale la plus fréquemment utilisée est l'hépaticojéjunostomie en Y de Roux. Cette procédure est habituellement effectuée par voie laparoscopique. Certaines études récentes ont observé que le résultat peut être amélioré par la chirurgie assistée par robot.
Pancréas annulaire
Le pancréas annulaire est une anomalie congénitale rare (de 5 à 15 par 100 000 naissances vivantes [5]), souvent associée au syndrome de Down, dans laquelle le tissu pancréatique encercle la 2e portion du duodénum, entraînant une occlusion duodénale.
Environ 2/3 des sujets touchés restent asymptomatiques. Parmi ceux qui développent des symptômes, les plus présents dans la période néonatale, mais la manifestation peut être retardée jusqu'à l'âge adulte. Les nouveau-nés présentent initialement des difficultés à s'alimenter et des vomissements qui peuvent être bilieux.
Le diagnostic de pancréas annulaire peut être suggéré par une rx de l'abdomen montrant le même signe de double bulle que celui observé dans l'atrésie duodénale. Le diagnostic peut également être posé par une opacification gastro-intestinale haute et est établi de façon plus précise par la TDM ou la cholangiopancréatographie IRM. La CPRE (cholangiopancréatographie rétrograde endoscopique) peut être effectuée chez les enfants plus âgés.
Le traitement du pancréas annulaire est le pontage chirurgical du pancréas annulaire avec duodénoduodénostomie, duodénojéjunostomie ou gastrojéjunostomie. La résection du pancréas doit être évitée en raison de complications potentielles liées à la pancréatite et au développement d'une fistule pancréatique.
Références
1. Bethell GS, Long AM, Knight M, Hall NJ; BAPS-CASS: Congenital duodenal obstruction in the UK: a population-based study. Arch Dis Child Fetal Neonatal Ed 105(2):178-183, 2020. doi:10.1136/archdischild-2019-317085
2. Engwall-Gill AJ, Zhou AL, Penikis AB, et al: Prenatal sonography in suspected proximal gastrointestinal obstructions: Diagnostic accuracy and neonatal outcomes. J Pediatr Surg 58(6):1090-1094, 2023. doi: 10.1016/j.jpedsurg.2023.02.029. Epub 2023 Feb 17. PMID: 36907770
3. Soares KC, Kim Y, Spolverato G, et al: Presentation and clinical outcomes of choledochal cysts in children and adults: a multi-institutional analysis. JAMA Surg 150(6):577-584, 2015. doi:10.1001/jamasurg.2015.0226
4. Søreide K, Søreide JA: Bile duct cyst as precursor to biliary tract cancer. Ann Surg Oncol 14(3):1200-1211, 2007. doi:10.1245/s10434-006-9294-3
5. Alkhayyat M, Bachour S, Abou Saleh M, et al: The epidemiology of annular pancreas in the United States: a population-based study. J Clin Gastroenterol 56(2):186-191, 2022. doi:10.1097/MCG.0000000000001531







