







La porphyrie cutanée tardive est une porphyrie hépatique relativement fréquente qui touche principalement la peau. L'atteinte du foie est également fréquente. La porphyrie cutanée tardive est due à un déficit acquis ou héréditaire de l'activité hépatique de l'uroporphyrinogène décarboxylase, une enzyme de la voie de biosynthèse de l'hème (voir tableau ). Les porphyrines s'accumulent, en particulier en cas d'augmentation du stress oxydatif dans les hépatocytes qui est généralement due à une augmentation du fer hépatique mais qui peut aussi être due à l'alcool, au tabagisme, aux œstrogènes, ou à l'hépatite C ou au VIH. Les symptômes comprennent une fragilité cutanée, des bulles qui se forment facilement sur la peau, principalement au niveau des zones exposées au soleil. Le diagnostic repose sur l'analyse des porphyrines dans les urines et les selles. La distinction avec la porphyrie cutanée aiguë (la coproporphyrie héréditaire et la porphyrie variegata) est importante. Le traitement comprend la déplétion en fer par saignée et l'augmentation de l'excrétion des porphyrines par l'administration de chloroquine ou d'hydroxychloroquine à faibles doses. La prévention consiste à éviter la lumière solaire, l'alcool, le tabac, les œstrogènes et les médicaments contenant du fer, et la réussite du traitement de toute infection par l'hépatite C et le VIH concomitantes.
(Voir aussi Revue générale des porphyries et Revue générale des porphyries cutanées.)
Physiopathologie de la porphyrie cutanée tardive
La porphyrie cutanée tardive (porphyrie cutanée tardive) résulte d'un déficit hépatique en uroporphyrinogène décarboxylase (UROD, voir tableau Substrats et enzymes de la voie de biosynthèse de l'hème). Les porphyrines s'accumulent dans le foie et sont transportées vers la peau, où elles favorisent une photosensibilité.
Les médicaments qui déclenchent généralement la porphyrie aiguë (voir le Drug Database for Acute Porphyria ou l'American Porphyria Foundation drug database) ne déclenchent pas la porphyrie cutanée tardive.
Une atteinte hépatique est fréquente dans la porphyrie cutanée tardive et peut être due en partie à l'accumulation des porphyrines, à une hépatite virale C chronique, à une hémosidérose concomitante ou à un abus d'alcool. Cirrhose survient chez ≤ 35% des patients et le carcinome hépatocellulaire survient chez 7 à 24% (plus fréquente parmi les hommes d’âge moyen).
Il existe deux types principaux de porphyrie cutanée tardive:
Type 1: acquis ou sporadique (75 à 80% des cas)
Type 2: héréditaire ou familial (20 à 25% des cas)
Il existe également un type 3 rare, qui explique < 1% des cas.
Dans la porphyrie cutanée tardive de type 1, la carence en décarboxylase est limitée au foie et aucune prédisposition génétique n’est présente. Cette porphyrie cutanée tardive se manifeste habituellement à un âge moyen ou plus avancé. Le déficit enzymatique est supposé être dû à l'augmentation du stress oxydatif dans les hépatocytes qui induit une oxydation accrue de l'uroporphyrinogène ou de l'uroporphyrine, qui n'est pas un substrat de l'enzyme, et à la formation de l'inhibiteur uroporphométhène. Les œstrogènes, l'excès de fer et l'infection par l'hépatite C augmentent également ce stress oxydatif dans les hépatocytes.
Dans la porphyrie cutanée tardive de type 2, le déficit en décarboxylase est héréditaire de manière autosomique dominante avec une pénétrance limitée. La carence se produit dans toutes les cellules, y compris les globules rouges. Elle se manifeste plus tôt que le type 1, parfois dans l'enfance. Le déficit (~50%) partiel en activité de l'uroporphyrinogène décarboxylase (UROD) chez les patients hétérozygotes ne suffit pas en soi à provoquer les caractéristiques biochimiques ou cliniques de la porphyrie cutanée tardive; d'autres facteurs sont nécessaires pour provoquer une diminution > 75% de l'activité de l'uroporphyrinogène décarboxylase (UROD) hépatique nécessaire pour que les signes de porphyrie cutanée tardive se manifestent. Ces facteurs peuvent comprendre une élévation du fer hépatique, une consommation d'alcool, une exposition aux hydrocarbures halogénés, le virus de l'hépatite C ou une infection par le VIH, la prise d'œstrogènes et le tabagisme. Ces facteurs augmentent l'oxydation des uroporphyrinogènes ainsi que d'autres porphyrinogènes en porphyrines correspondantes et facilitent aussi la formation d'inhibiteurs de l'uroporphyrinogène décarboxylase (UROD).
La porphyrie hépatoérythropoïétique (voir tableau Quelques porphyries moins fréquentes), qui correspond à une carence profonde en UROD, est très rare et est souvent considérée comme une forme autosomique récessive de porphyrie cutanée tardive de type 2.
La porphyrie cutanée tardive de type 3, est très rare, et est héréditaire, mais sans aucun défaut en UROD; un défaut d'un autre gène, non identifié semble en cause.
Les types 1 et 2 sont les principales formes de porphyrie cutanée tardive. Elles ont les mêmes facteurs déclenchants, les mêmes symptômes et le même traitement. La prévalence globale peut être de l'ordre de 1/10 000, mais est probablement plus élevée chez les sujets exposés à des hydrocarbures aromatiques halogénés ou à d'autres facteurs déclenchants de la maladie.
Pseudoporphyrie
L'insuffisance rénale terminale, le rayonnement ultraviolet (UVA) et certains médicaments peuvent provoquer des symptômes similaires à ceux des porphyries cutanées tardives sans taux de porphyrines élevés (pseudoporphyrie). Les médicaments couramment impliqués sont le furosémide, les tétracyclines, les sulfamides, et le naproxène et d'autres AINS.
Les porphyrines dialysant mal, certains patients sous hémodialyse au long cours développent des lésions cutanées qui ressemblent à la porphyrie cutanée tardive (pseudo-porphyrie de néphropathie terminale); cette pathologie est appelée pseudo-porphyrie de la maladie rénale terminale.
Symptomatologie de la porphyrie cutanée tardive
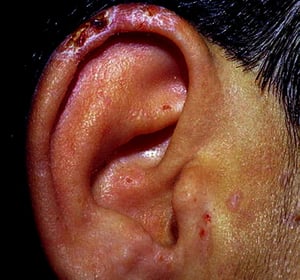
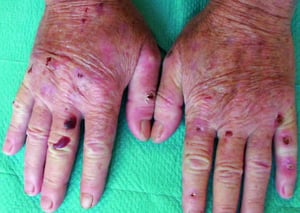
Le patient qui souffre de porphyrie cutanée tardive se présente avec une fragilité cutanée, principalement des zones exposées au soleil. La phototoxicité est retardée: par conséquent, le patient ne fait pas toujours le rapport entre l'exposition solaire et les symptômes.
© Springer Science+Business Media
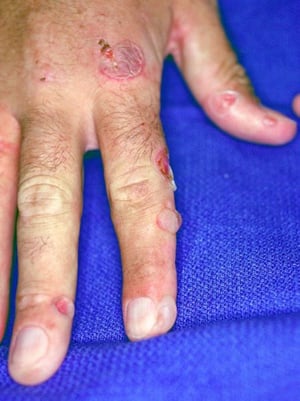
Image courtesy of Karen McKoy, MD.
By permission of the publisher. D'après White K, Soter N. In Current Dermatologic Diagnosis and Treatment. Edited by I Freedberg, IM Freedberg, and MR Sanchez. Philadelphia, Current Medicine, 2001.
© Springer Science+Business Media


© Springer Science+Business Media


Image courtesy of Karen McKoy, MD.


By permission of the publisher. D'après White K, Soter N. In Current Dermatologic Diagnosis and Treatment. Edited by I Freedberg, IM Freedberg, and MR Sanchez. Philadelphia, Current Medicine, 2001.


© Springer Science+Business Media
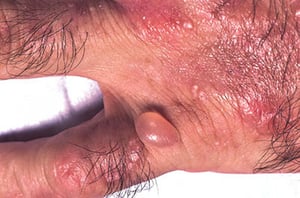
Spontanément ou après un traumatisme mineur, des bulles tendues se développent. Certaines bulles sont hémorragiques. Les érosions et ulcérations qui les accompagnent peuvent s’infecter secondairement; elles cicatrisent lentement, laissant des escarres atrophiques. L'exposition solaire conduit parfois à une érythrose, un œdème ou un prurit.
Une conjonctivite hyperhémique peut se développer, mais les autres muqueuses ne sont pas affectées.
Des zones d'hypopigmentation ou d'hyperpigmentation peuvent apparaître, de même qu'une hypertrichose faciale et des lésions pseudo-sclérodermiques.
Diagnostic de la porphyrie cutanée tardive
Taux élevés de porphyrines plasmatiques, de l'uroporphyrine et de l'heptacarboxylporphyrine urinaires, et de l'isocoproporphyrine fécale
Chez un patient par ailleurs sain, la fragilité cutanée et la formation de phlyctènes sont évocatrices de la porphyrie cutanée tardive. La distinction avec les porphyries aiguës avec symptômes cutanés (porphyrie variegata et coproporphyrie héréditaire) est importante parce qu’en cas de porphyrie variegata et de coproporphyrie héréditaire, la prescription erronée de médicaments porphyrogènes peut déclencher les symptômes neuroviscéraux graves des porphyries aiguës. Des symptômes neurologiques ou des antécédents de couleurs abdominales peuvent suggérer une porphyrie aiguë. Les antécédents d'exposition à des produits chimiques qui peuvent favoriser la pseudo-porphyrie doivent être recherchés.
Bien que toutes les porphyries qui entraînent des lésions cutanées, une élévation des porphyrines plasmatiques, une élévation de l'uroporphyrine, de l'heptacarboxylporphyrine urinaires et de l'isocoproporphyrine fécale soient en faveur de la porphyrie cutanée tardive. Les taux urinaires de porphobilinogène précurseur de porphyrine (PBG) sont normaux dans la porphyrie cutanée tardive. L'acide delta-aminolévulinique urinaire peut être légèrement augmenté (< 3 fois la limite supérieure de la normale). L’activité érythrocytaire de l'uroporphyrinogène décarboxylase (UROD) est normale dans la porphyrie cutanée tardive de type 1 et de type 3, mais est diminuée (d'environ 50%) dans le type 2.
L'hépatite C et le VIH doivent être recherchés chez les patients qui présentent une porphyrie cutanée tardive (1). Une surcharge en fer doit être recherchée par la mesure du fer sérique, du taux de ferritine, et de la capacité de fixation totale du fer; si les examens sont en faveur d'une surcharge en fer, des tests génétiques à la recherche d'une mutation du gène HFE de l'hémochromatose héréditaire seront pratiqués (2).
Références pour le diagnostic
1. Caballes FR, Sendi H, Bonkovsky HL: Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int 32(6):880–893, 2012. doi:10.1111/j.1478-3231.2012.02794.x
2. Bonkovsky HL, Guo JT, Hou W, et al: Porphyrin and heme metabolism and the porphyrias. Compr Physiol 3(1):365–401, 2013. doi:10.1002/cphy.c120006
Traitement de la porphyrie cutanée tardive
Quatre stratégies thérapeutiques complémentaires sont disponibles:
Réduction de dépôts tissulaires de fer
Augmentation de l'excrétion des porphyrines
Traitement de l'hépatite C chronique et de l'infection par le VIH, le cas échéant
Arrêt de la consommation d'alcool, du tabagisme et de la consommation d'œstrogènes, le cas échéant
Ces stratégies peuvent être combinées en vue d'une rémission plus rapide bien que leur association ne soit généralement pas nécessaire. Le traitement est surveillé par la détermination de la ferritinémie (en cas de traitement de réduction du fer) et des taux d'excrétion urinaire des porphyrines tous les 2 ou 3 mois jusqu'à rémission complète.
Les patients doivent arrêter de consommer de l'alcool et de fumer (y compris la marijuana).
L'élimination du fer par des saignées est habituellement efficace. Une unité de sang est retirée chaque semaine ou deux. Lorsque la ferritinémie sérique descend légèrement en deçà de la normale, les saignées sont interrompues. Habituellement, 6 à 10 séances sont nécessaires. Les porphyrines plasmatiques et urinaires diminuent progressivement sous traitement, un peu en retard mais parallèlement à la diminution de la ferritine. L'aspect de la peau finit par se normaliser. Après la rémission, des saignées ultérieures ne sont nécessaires qu'en cas de récidive. Un traitement continu de réduction du fer est également recommandé en cas d'hémochromatose héréditaire. Chez ces patients, la ferritine sérique cible est de 50 à 150 ng/mL (50 à 150 microgrammes/L).
La chloroquine à faible dose ou l'hydroxychloroquine 100 à 125 mg par voie orale 2 fois/semaine diminuent l'excès des porphyrines dans le foie et peut-être d'autres tissus en augmentant leur taux d'excrétion. Des doses plus importantes peuvent favoriser des lésions hépatiques transitoires et l'aggravation de la porphyrie. Lorsque la rémission est obtenue, le traitement est arrêté.
La chloroquine et l'hydroxychloroquine sont inefficaces dans les néphropathies avancées et les saignées sont habituellement contre-indiquées du fait de l'anémie sous-jacente. Cependant, l'érythropoïétine recombinante mobilise l'excès de fer et améliore suffisamment l'anémie pour permettre la saignée. En phase terminale, la déféroxamine est un complément à la saignée pour réduire le fer hépatique, le fer complexé étant enlevé par la dialyse. Les dialyseurs avec les membranes ultra-perméable et très haute débits sanguins sont nécessaires.
Les patients qui ont une porphyrie cutanée tardive évidente et une infection par l'hépatite C doivent être évalués pour un traitement par des médicaments antiviraux à action directe. Les antécédents de déplétion en fer augmentent la réponse au traitement antiviral à base d'interféron, mais cette déplétion ne semble pas importante car le traitement actuel utilise des médicaments antiviraux à action directe très efficaces. Une rémission des lésions cutanées de la porphyrie cutanée tardive après traitement antiviral a été rapportée, mais la documentation de l'inversion complète des défauts métaboliques et de l'amélioration à long terme fait défaut. Néanmoins, il semble utile de traiter et de guérir l'infection par le virus de l'hépatite C avant de décider de la réduction du fer ou d'un traitement par l'hydroxychloroquine chez ces patients (1). L'infection par le VIH doit être traitée de manière appropriée tout en traitant la porphyrie cutanée tardive.
Les enfants présentant une porphyrie cutanée tardive symptomatique sont traités par des petites saignées ou par chloroquine orale; la posologie est déterminée suivant le poids corporel.
Les symptômes cutanés survenant pendant la grossesse sont traités par des saignées. Dans les cas réfractaires, de faibles doses de chloroquine peuvent être ajoutées; aucun effet tératogène n’a été reconnu. Suivant le degré d'hémodilution et la carence en fer, les symptômes cutanés s'atténuent habituellement avec l'avancée de la grossesse.
La supplémentation en œstrogènes après la ménopause ou d'autres traitements œstrogéniques, tels que chez les hommes atteints d'un cancer de la prostate, est interrompue pendant le traitement de la porphyrie cutanée tardive. L'arrêt des œstrogènes induit souvent la rémission. La substitution des œstrogènes systémiques par des œstrogènes transdermiques est parfois effectuée si les symptômes de la ménopause sont gênants, mais le risque d'absorption systémique peut persister.
La cimétidine a été suggérée comme traitement de la porphyrie cutanée tardive, car on estime que la cimétidine régule négativement l'ALA synthétase hépatique-1. Cependant, l'ALA synthétase hépatique-1 n'est pas mesurée augmentée dans la porphyrie cutanée tardive. Ainsi, l'utilisation systématique de la cimétidine dans la prise en charge de la porphyrie cutanée tardive n'est pas recommandée (2).
Références pour le traitement
1. Rudnick S, Bonkovsky HL: Hepatitis C and porphyria cutanea tarda in 2020. Aliment Pharmacol Therap 51:1432–1434, 2020. doi: 10.1111/apt.15728
2. Fujita Y, Sato-Matsumura KC: Effective treatment for porphyria cutanea tarda with oral cimetidine. J Dermatol 37(7):677–679, 2010. doi:10.1111/j.1346-8138.2010.00838.x
Prévention de la porphyrie cutanée tardive
Les patients doivent éviter l'exposition au soleil, en utilisant des chapeaux et des vêtements et des écrans solaires en oxyde de zinc ou de titane. Les écrans solaires classiques qui bloquent les UV sont inefficaces, mais les écrans solaires absorbant les UVA comme ceux contenant des dibenzoylméthanes permettent d'obtenir une protection un peu meilleure. L'ingestion d'alcool doit être évitée définitivement et le tabagisme doit être arrêté. La supplémentation en œstrogènes, en particulier administrée par voie transdermique à faible dose, peut généralement être reprise en toute sécurité après une rémission de la maladie.
Points clés
La porphyrie cutanée tardive est généralement acquise, mais peut être héréditaire.
Les déclencheurs comprennent un fer hépatique élevé, la consommation d'alcool, l'exposition aux hydrocarbures halogénés, et l'infection par le virus de l'hépatite C ou par le VIH.
Les médicaments qui déclenchent généralement la porphyrie aiguë ne déclenchent pas la porphyrie cutanée tardive.
Mesurer les porphyrines et l'heptacarboxyl porphyrine urinaires et l'isocoproporphyrine fécale.
Tester la surcharge en fer avec les taux de fer et de ferritine sériques et la capacité totale de fixation du fer.
Tester les patients qui ont une ferritine sérique ou une saturation de la transferrine élevées à la recherche de mutations génétiques d'HFE.
Réduire les stocks élevés de fer par des saignées.
Éliminer les porphyrines en excès en administrant de la chloroquine ou de l'hydroxychloroquine à faible dose.
Traiter les patients atteints d'hépatite C par des médicaments antiviraux à action directe.
Plus d'information
Les sources d'information suivantes en anglais peuvent être utiles. S'il vous plaît, notez que LE MANUEL n'est pas responsable du contenu de ces ressources.
American Porphyria Foundation: vise à éduquer et à soutenir les patients et les familles touchés par les porphyries et à soutenir la recherche sur le traitement et la prévention des porphyries
American Porphyria Foundation: Safe/Unsafe Drug Database: Provides an up-to-date list of medications available in the United States to assist physicians in prescribing for patients with porphyrias
United Porphyrias Association: fournit une formation et un soutien aux patients et à leurs familles; fournit des informations fiables aux professionnels de santé; favorise et soutient la recherche clinique pour améliorer le diagnostic et la prise en charge des porphyries
European Porphyria Network: Promotes clinical research about porphyrias
The Drug Database for Acute Porphyrias: Provides an up-to-date list of medications available in Europe to assist physicians in prescribing for patients with porphyrias







