







Les tumeurs osseuses malignes primitives sont beaucoup moins fréquentes que les tumeurs osseuses métastatiques, en particulier chez l'adulte. Les tumeurs malignes osseuses primitives comprennent le myélome multiple, l'ostéosarcome, l'adamantinome, le chondrosarcome, le chordome, le sarcome d'Ewing osseux, le fibrosarcome et le sarcome pléomorphe indifférencié, le lymphome osseux et les tumeurs malignes à cellules géantes. (Voir aussi Revue générale des tumeurs osseuses et articulaires et Revue générale des leucémies.)
Les deux systèmes les plus utilisés pour classer ces tumeurs sont
L'American Joint Committee on Cancer (AJCC) Cancer Staging Manual, 8th edition: dans le cas de l'ostéosarcome, du chondrosarcome et du sarcome d'Ewing, la classification par stade est basée la catégorie, le grade histologique, la taille tumorale, l'atteinte ganglionnaire et les métastases (classification TNM). Le manuel classe les tumeurs en 4 stades et est utilisé pour rapporter les données sur le cancer.
Le système de classification par stade de la Musculoskeletal Tumor Society (MSTS): utilisé par les chirurgiens orthopédistes oncologues basé sur le grade histologique (p. ex., stade I-histologie de bas grade et stade II-histologie de haut grade, si la tumeur est entièrement contenue dans l'os (A) ou s'est propagée à l'extérieur du cortex dans les tissus mous environnants (B) et métastases, stade III). L'ostéosarcome typique (conventionnel) avec une masse de tissus mous associée sans métastases est le stade IIB du système MSTS.
Myélome multiple
Le myélome multiple est la plus fréquente des tumeurs osseuses malignes primitives, mais il est souvent considéré comme une tumeur des cellules de la moelle osseuse plutôt qu'une tumeur osseuse primitive car il s'agit d'une dérivation hématopoïétique (voir aussi Myélome multiple). Bien que le myélome multiple soit considéré comme une tumeur hématologique, l'anomalie squelettique identifiée doit être différenciée des autres tumeurs osseuses.
Le myélome multiple se produit principalement chez les personnes âgées.
Le développement et la progression tumorale sont habituellement multicentriques et touchent la moelle osseuse souvent de façon si diffuse que le myélogramme permet le diagnostic. Contrairement à ce que l'on observe dans la maladie métastatique, une scintigraphie osseuse peut ne pas montrer de manière fiable les lésions; des examens complémentaires du squelette doivent être effectués. Ces examens squelettiques montrent généralement des lésions lytiques bien circonscrites (lésions à l'emporte-pièce) ou une déminéralisation diffuse. Rarement, la lésion peut apparaître comme condensée ou comme une ostéopénie diffuse, en particulier dans un corps vertébral. Une lésion myélomateuse unique, sans atteinte de la moelle systémique est appelée plasmocytome.

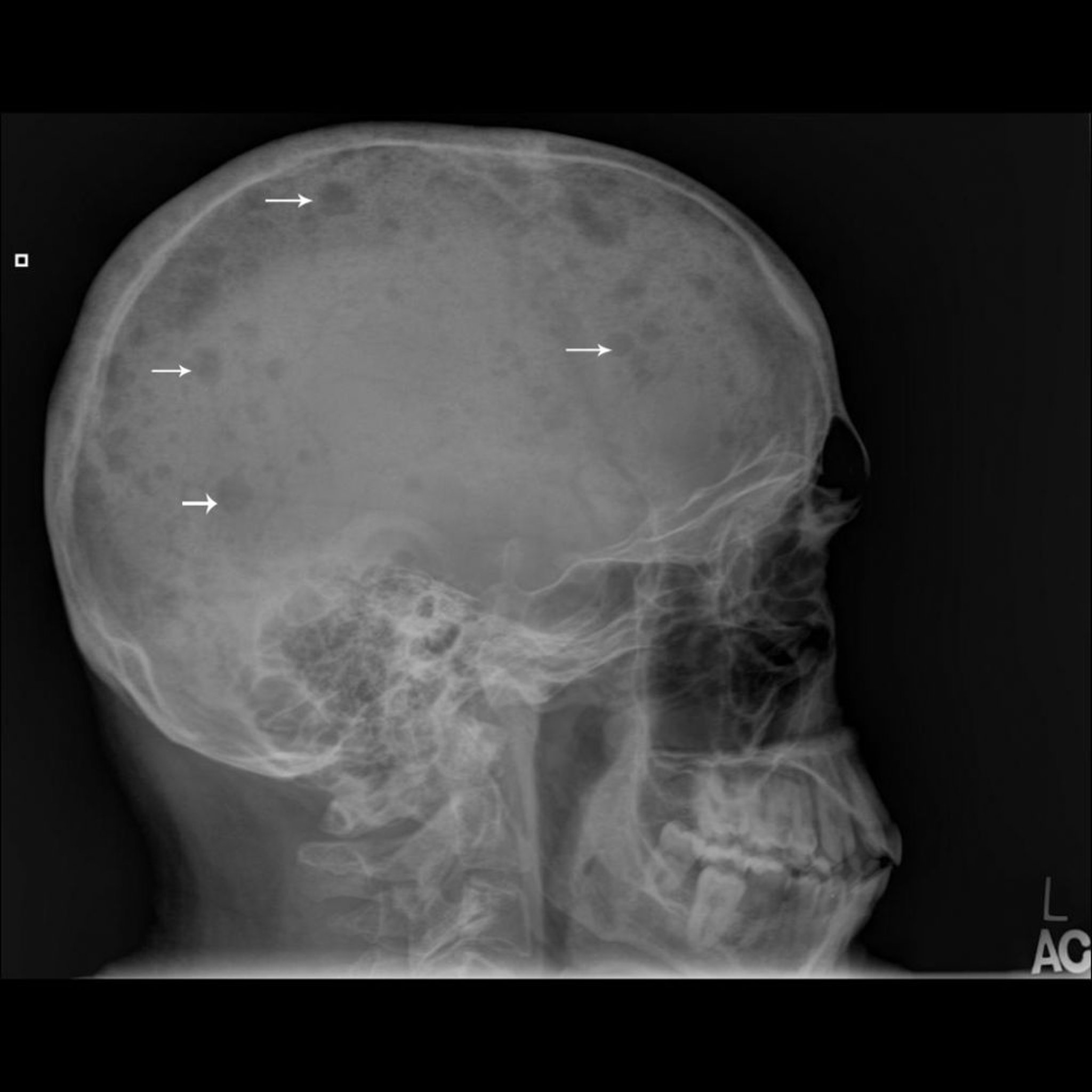
Image courtesy of Michael J. Joyce, MD, et Hakan Ilaslan, MD.
Certaines lésions osseuses répondent assez bien à la radiothérapie.
Ostéosarcome (sarcome ostéogénique)
L'ostéosarcome est la tumeur osseuse primitive maligne la plus fréquente (si l'on considère le myélome comme une tumeur des cellules médullaires et non comme une tumeur osseuse primitive) et est très malin. Il est plus fréquent chez les sujets âgés de 10 à 25 ans, mais il peut survenir à tout âge. Il y a deux pics d'incidence; l'incidence est la plus élevée chez les adolescents et les très jeunes adultes (ce qui coïncide avec la poussée de croissance de l'adolescence) et un pic secondaire chez les personnes âgées (≥ 60 ans), en particulier chez celles présentant des facteurs de risque tels qu'une maladie de Paget, des infarctus osseux et de zones osseuses précédemment exposées à une radiothérapie à haute dose pour un autre cancer plusieurs années auparavant. Il existe une prédisposition génétique, en particulier chez l'enfant porteur du gène du rétinoblastome héréditaire (variantes du gène RB1) et du syndrome de Li-Fraumeni (gène TP53).
Les cellules osseuses tumorales de l'ostéosarcome produisent un os ostéoïde, immature. L'ostéosarcome se développe généralement autour du genou (fémur distal plus souvent que tibia proximal) ou dans d'autres os longs, en particulier dans la région métaphysodiaphysaire, et peut métastaser, généralement au poumon ou dans un autre os. La douleur et la tuméfaction sont les symptômes habituels.

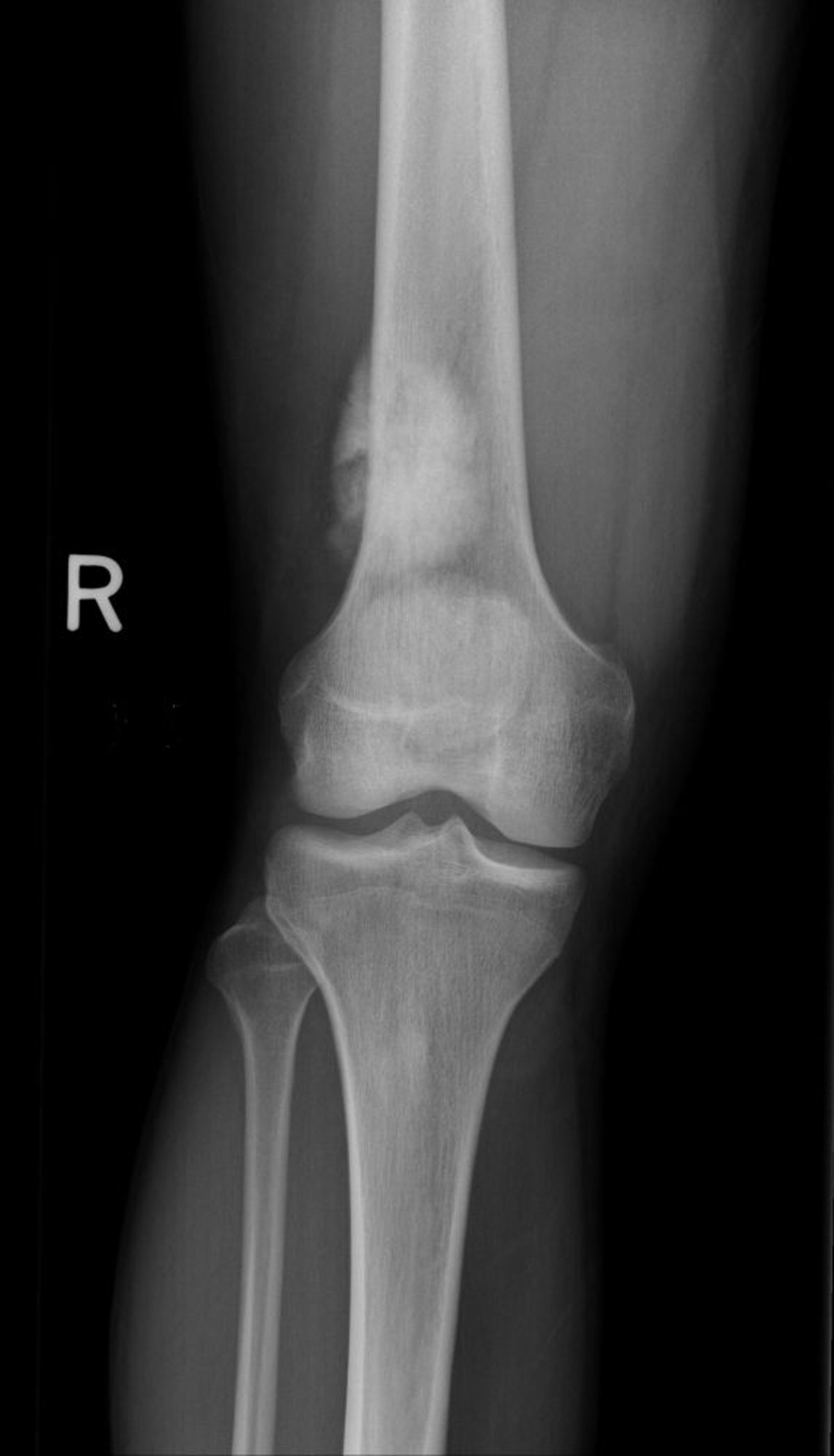
Image courtesy of Michael J. Joyce, MD, et Hakan Ilaslan, MD.
Les données de l'imagerie varient et peuvent inclure des aspects condensés ou lytiques. Le diagnostic d'ostéosarcome nécessite une biopsie. Les patients doivent subir une rx thorax et d'une TDM pour détecter des métastases pulmonaires et une scintigraphie osseuse pour détecter des métastases osseuses. L'IRM est réalisée sur l'ensemble de l'extrémité concernée pour détecter les lésions métachrones si présentes. La TEP-TDM peut montrer des métastases à distance ou des lésions métachrones.
Le traitement de l'ostéosarcome est basé sur une association de chimiothérapie et de chirurgie. L'utilisation de la chimiothérapie adjuvante accroît la survie de < 20% à > 65% à 5 ans. La chimiothérapie néoadjuvante commence avant la résection chirurgicale. La diminution de la masse tumorale des tissus mous périphériques ou l'augmentation de la minéralisation sur les radiographies ont diminué le niveau de douleur, et une diminution du taux de la phosphatase alcaline sérique indiquent une réponse au traitement, mais la réponse souhaitée est de > 95% de nécrose tumorale sur la cartographie histologique de la pièce de résection observée par l'anatomopathologiste. Après plusieurs cures de chimiothérapie (sur plusieurs mois) la chirurgie épargnant le membre et la chirurgie de reconstruction du membre peuvent prendre place. À l'occasion, une amputation chirurgicale est effectuée avant le début de la chimiothérapie pour une tumeur fongique. L'objectif est de traiter la maladie micrométastatique précoce supposée être présente même si elle n'est pas observée sur l'imagerie de définition du stade.
Dans la chirurgie conservatrice, la tumeur est réséquée en bloc, y compris tous les tissus environnants réactionnels ainsi qu'une berge de tissu normal autour de la tumeur; pour éviter les fuites microscopiques de cellules tumorales, la tumeur n'est pas ouverte avant d'être extraite. Plus de 85% des patients peuvent être traités par la chirurgie orthopédique conservatrice sans diminution du taux de survie à long terme.
Il est nécessaire de poursuivre la chimiothérapie après la chirurgie. Si la nécrose tumorale est presque complète (environ 95%) après la chimiothérapie pré-opératoire, le taux de survie à 5 ans est > 90%. Une maladie métastatique limitée aux poumons peut parfois être traitée par thoracotomie et résection en coin (cunéiforme ou résection wedge) des lésions pulmonaires.
Des variantes de l'ostéosarcome différentes de l'ostéosarcome classique qui se produisent beaucoup moins fréquemment comprennent des lésions superficielles corticales, comme l'ostéosarcome parostéal et l'ostéosarcome périosté. L'ostéosarcome parostéal (ou parostéal) atteint le plus souvent la corticale postérieure du fémur distal et est généralement assez bien différencié. La chimiothérapie n'est pas nécessaire avant la résection chirurgicale dans le traitement de l'ostéosarcome parostéal de bas grade. L'ostéosarcome parostéal nécessite une résection chirurgicale en bloc, mais ne relève pas de la chimiothérapie si l'histologie du prélèvement réséqué confirme que la tumeur est bien différenciée.
L'ostéosarcome périosté est plutôt une tumeur de la surface de la matrice cartilagineuse qui contient également une matrice osseuse et est malin. Il est souvent situé au milieu du fut fémoral et apparaît comme un bijou en forme de soleil sur les rx. La probabilité de métastases à partir de l'ostéosarcome périosté est beaucoup plus élevée que pour l'ostéosarcome parostéal bien différencié, mais un peu plus faible que pour les ostéosarcomes typiques. La plupart du temps, les ostéosarcomes périostés sont traités comme les ostéosarcomes conventionnels par chimiothérapie et résection chirurgicale en bloc.
Adamantinome
L'adamantinome est rare (< 1% des tumeurs malignes des os) et se développe le plus souvent dans le tibia. Il est le plus souvent observé chez les adolescents et entre 20 et 30 ans, mais peut survenir à tout âge. L'adamantinome a une croissance lente et se manifeste souvent par une douleur et une plénitude palpable.

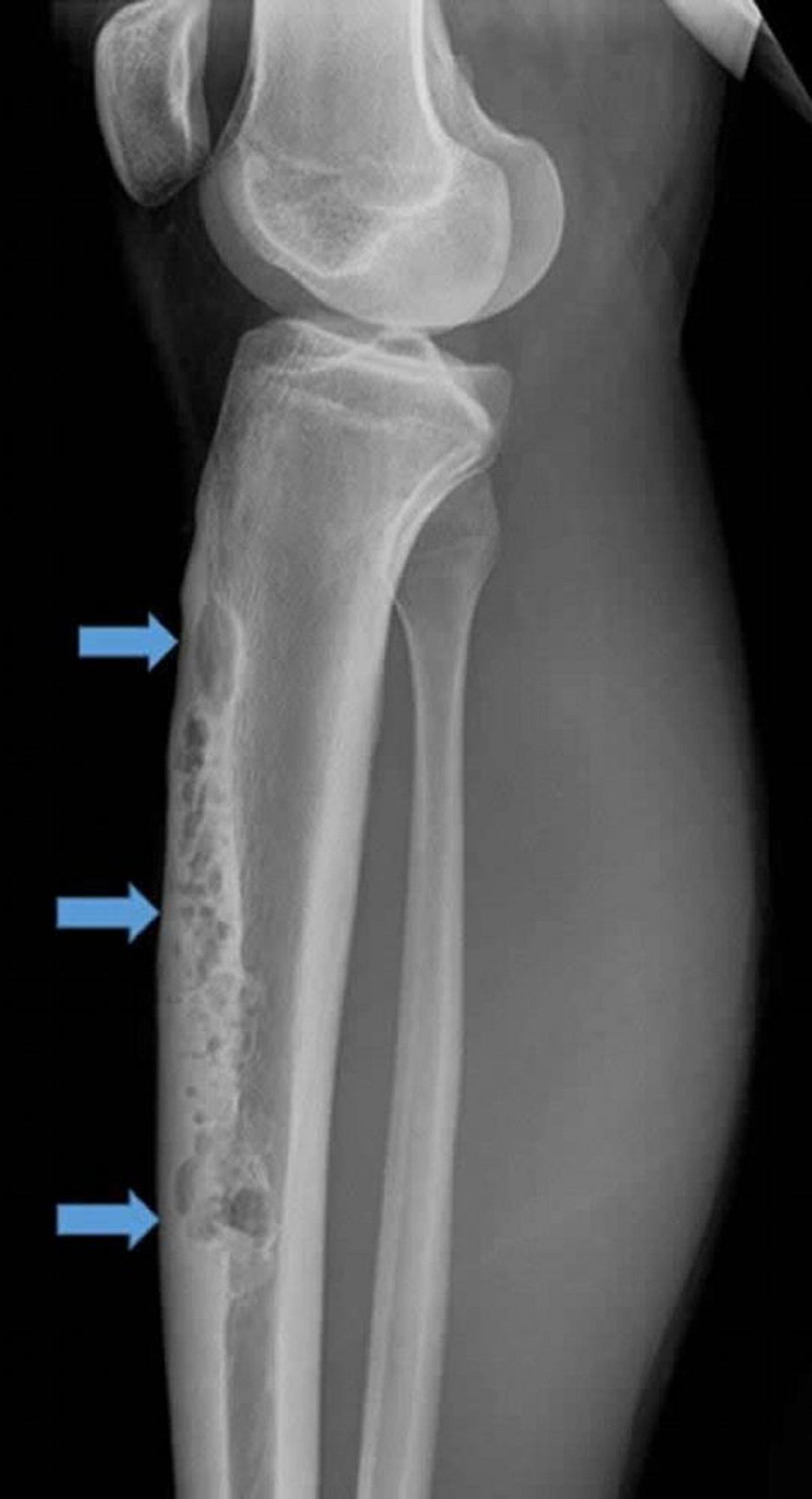
Image courtesy of Michael J. Joyce, MD, et Hakan Ilaslan, MD.
La lésion se manifeste généralement au niveau de la crête antérieure du tibia et les rx montrent un aspect ostéolytique en "bulle de savon". L’aspect histologique est un motif biphasique de tissu épithélial et ostéofibreux. La lésion peut être confondue avec une dysplasie ostéofibreuse du cortex tibial antérieur, qui est bénigne. Certains pensent que la dysplasie ostéofibreuse du cortex tibial antérieur pourrait être un précurseur de l'adamantinome, mais sans la composante épithéliale qui en ferait alors un cancer.
Les métastases sont principalement aux poumons, mais sont rares.
Le traitement de l'adamantinome consiste en une résection large et en une reconstruction du défect. Une amputation est parfois nécessaire.
Chondrosarcome
Les chondrosarcomes sont des tumeurs malignes du cartilage. Ils diffèrent des ostéosarcomes sur les plans clinique, thérapeutique et pronostique. Quatre-vingt-dix pour cent des chondrosarcomes sont des tumeurs primitives. La survenue d'un chondrosarcome peut également émailler l'évolution d'autres troubles préexistants, en particulier les ostéochondromes multiples et l'enchondromatose multiple (p. ex., dans la maladie d'Ollier et dans le syndrome de Maffucci). Les chondrosarcomes ont tendance à survenir chez les personnes âgées. Ils se développent souvent dans les os plats (p. ex., le bassin, l'omoplate), mais peuvent se développer dans n'importe quelle partie de n'importe quel os (le plus souvent le fémur et l'humérus parmi les os longs) et peut avoir une composante tumorale des tissus mous impliquant les tissus mous environnants.

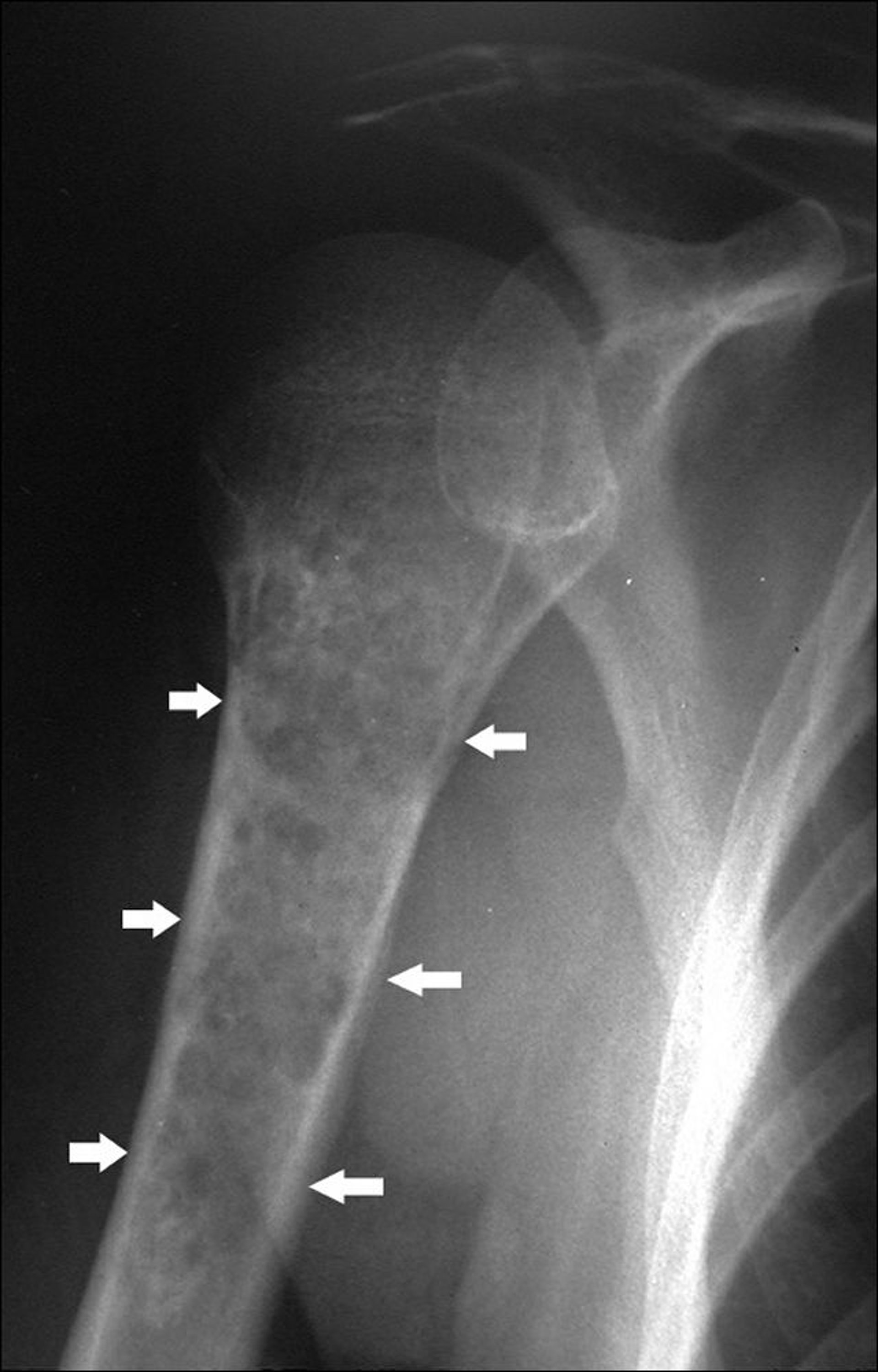
Image courtesy of Michael J. Joyce, MD, et Hakan Ilaslan, MD.
Les rx révèlent souvent des calcifications punctiformes. Les chondrosarcomes donnent également souvent une destruction de l'os cortical et une perte des travées osseuses normales. L'IRM peut montrer une masse dans les tissus mous. Une scintigraphie osseuse peut également être effectuée. Le diagnostic tissulaire est nécessaire au diagnostic de chondrosarcome et peut également déterminer le grade de la tumeur (qui conditionne la probabilité de métastases). La biopsie à l'aiguille peut fournir un échantillon de tissu inadéquat.
Il est souvent difficile de différencier les chondrosarcomes de bas grade des enchondromes par l'imagerie et parfois même par l'histologie.
Les chondrosarcomes de bas grade (grade 1) sont souvent traités par curetage large intralésionnel associé à un traitement adjuvant (souvent la congélation par l'azote liquide, ou l'irradiation à l'argon, ou la chaleur du méthacrylate de méthyle, ou la radiofréquence, ou encore le phénol). Certains chirurgiens préfèrent la résection chirurgicale en bloc dans le cas des tumeurs de bas grade afin de réduire le risque de récidive. Les tumeurs de grade plus élevé sont traitées par résection chirurgicale en bloc. Lorsque la résection chirurgicale avec maintien de la fonction est impossible, l'amputation peut être nécessaire. En raison de la possibilité d'une contamination tumorale, un soin méticuleux est de mise pour éviter les fuites de cellules tumorales dans les tissus mous lors d'une biopsie ou de la chirurgie. La récidive est inévitable si des cellules tumorales se répandent hors de la tumeur. Si aucune fuite ne se produit, le taux de guérison dépend du grade tumoral. Les tumeurs de bas grade sont presque toutes guéries par un traitement adéquat. Comme ces tumeurs sont peu vascularisées, la chimiothérapie et la radiothérapie ont peu d'efficacité.
Chordome
Le chordome, qui est rare, se développe à partir des restes de la notochorde primitive. Il affectionne les extrémités de la colonne vertébrale, en général au milieu du sacrum ou près de la base du crâne. Un chordome dans la région sacrococcygienne provoque une douleur presque constante. Un chordome dans la base du crâne peut causer des déficits dans un nerf crânien, le plus souvent dans les nerfs de l'œil.
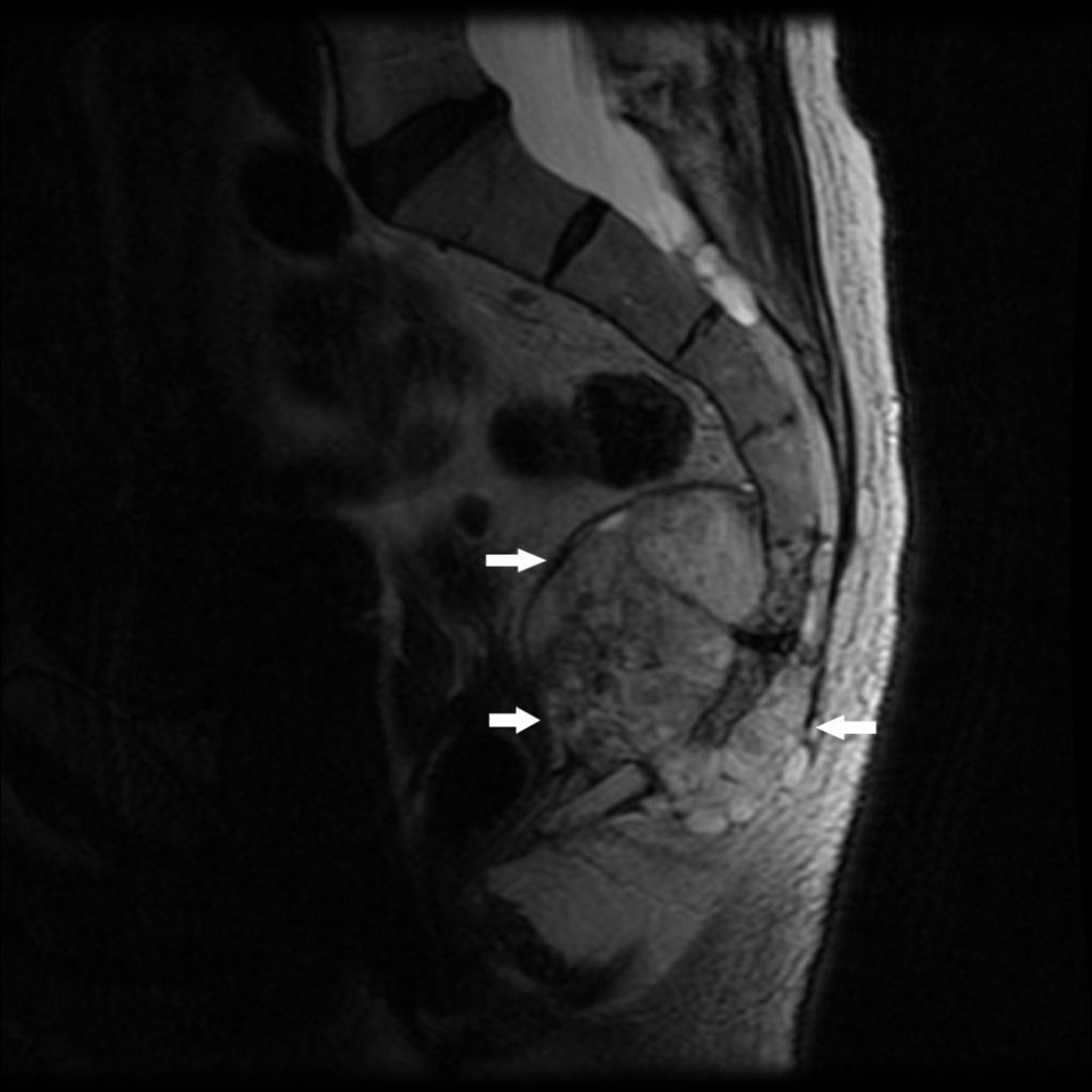
Image courtesy of Michael J. Joyce, MD, et Hakan Ilaslan, MD.
Les symptômes du chordome peuvent précéder le diagnostic de plusieurs mois voire des années.
Un chordome apparaît sur l'imagerie comme une lésion osseuse destructrice qui peut être associée à une masse de tissus mous. Une biopsie est effectuée.
Les chordomes de la région sacrococcygienne peuvent être guéris par l'excision radicale en bloc. Les chordomes de la base du crâne sont généralement inaccessibles à la chirurgie mais peuvent répondre à la radiothérapie. Le taux de récidive locale est élevé après résection de la tumeur. Des métastases, bien que moins fréquentes, peuvent survenir.
Sarcome d'Ewing de l'os
Le sarcome d'Ewing de l'os est une tumeur osseuse à cellules rondes bleue avec un pic d'incidence entre 10 et 20 ans. Le sarcome d'Ewing est lié aux tumeurs neuroectodermiques périphériques primitives (peripheral primitive neuroectodermal tumors, PNET) et aux tumeurs malignes à petites cellules d'Askin de la paroi thoracique, qui sont à présent considérées comme faisant partie de la famille des sarcomes d'Ewing. La plupart des tumeurs se développent dans les extrémités, mais n'importe quel os peut être atteint. Le sarcome d'Ewing a tendance à se répandre, envahissant parfois tout l'os, le plus souvent la région diaphysaire. Environ 15 à 20% surviennent dans la région métaphysaire. La douleur et la tuméfaction sont les symptômes les plus fréquents.

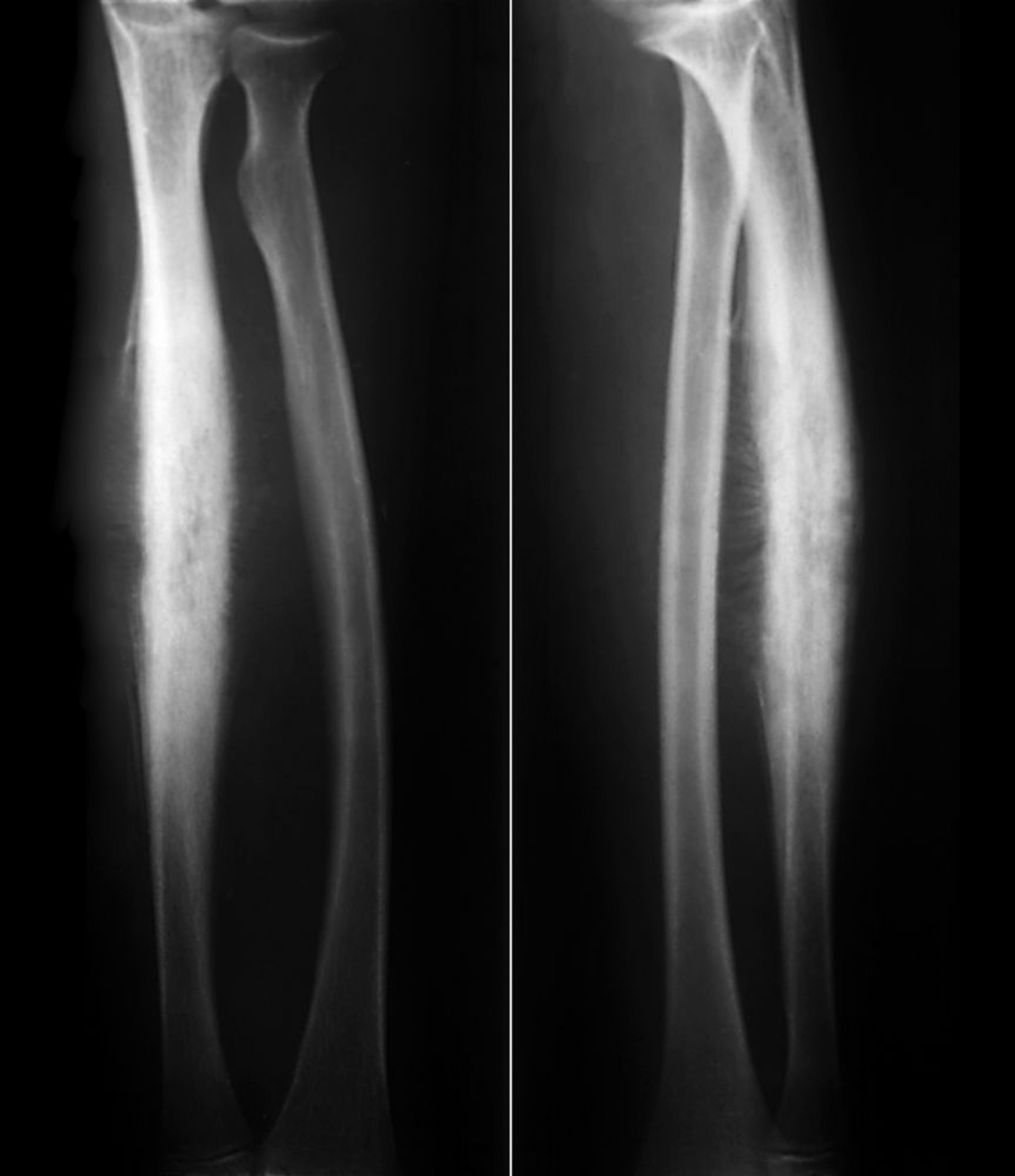
ZEPHYR/SCIENCE PHOTO LIBRARY
La lyse osseuse, en particulier sur un mode infiltratif avec perméation sans bords précis, est l'image la plus fréquente sur l'imagerie, mais de multiples couches de néoformation osseuse réactionnelle sous-périostée nouvelle peuvent donner un aspect de peau d'oignon. La rx ne peut généralement pas évaluer l'étendue de l'atteinte osseuse, et une grande masse de tissus mous entoure habituellement l'os touché. L'IRM définit mieux l'importance du processus, ce qui permet d'orienter le traitement.
Beaucoup d'autres tumeurs bénignes et malignes peuvent apparaître de façon très similaire, aussi le diagnostic du sarcome d'Ewing repose-t-il sur la biopsie. Parfois, ce type de tumeur peut être confondu avec une infection. Le diagnostic histologique précis peut être fait avec analyse cytogénétique et identification des marqueurs moléculaires, notamment la recherche d'un type d'anomalie chromosomique clonale. Les signes moléculaires de la famille des tumeurs d'Ewing sont des translocations chromosomiques non aléatoires particulières impliquant le gène du sarcome d'Ewing (Ewing sarcoma gene [EWS]) sur le chromosome 22. Dix-huit translocations structurelles différentes avec différents modèles de gènes de fusion ont été identifiées.

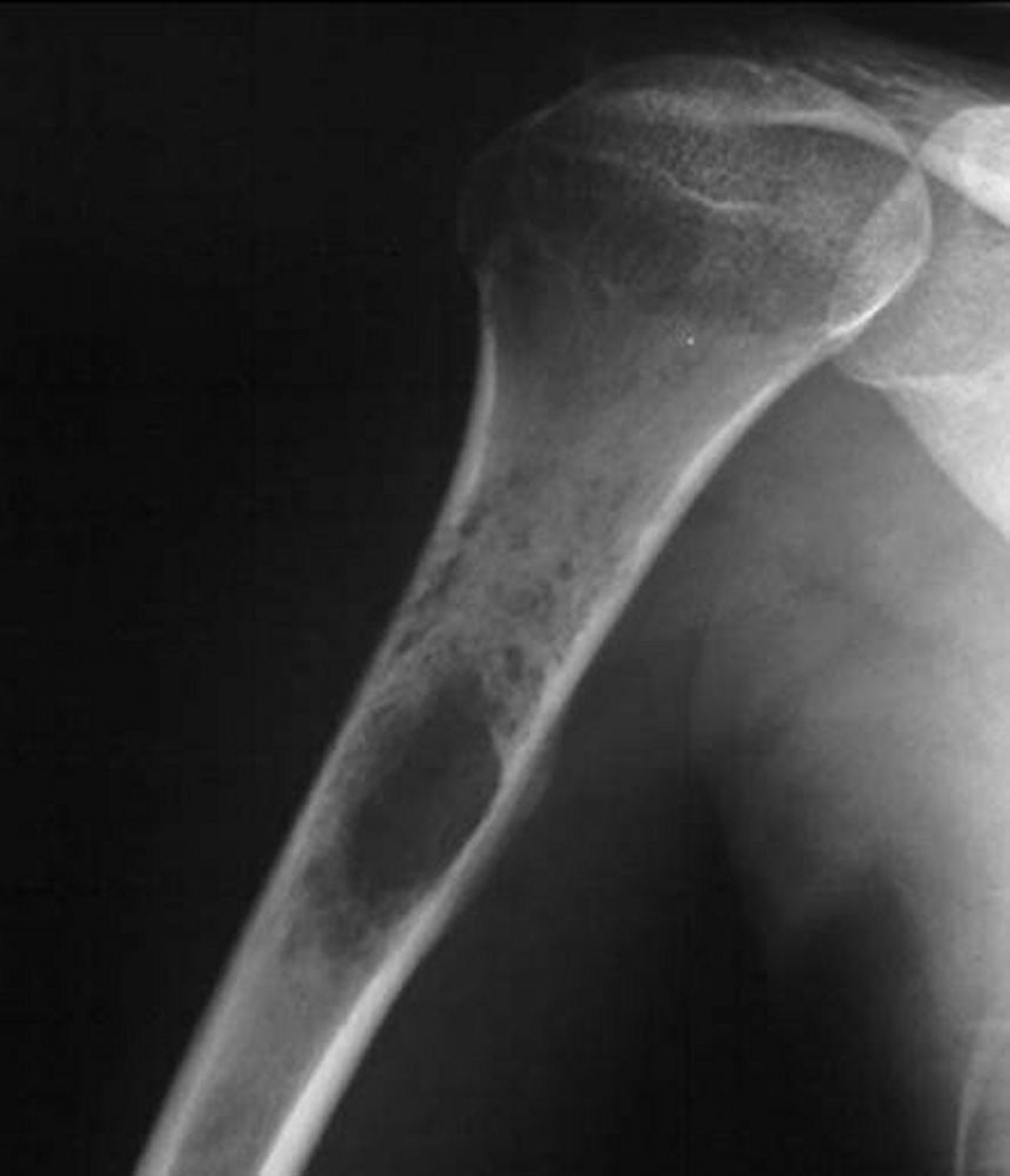
Image courtesy of Michael J. Joyce, MD, et Hakan Ilaslan, MD.
Le traitement du sarcome d'Ewing comprend diverses associations de chirurgie, de chimiothérapie et de radiothérapie. Actuellement, > 60% des patients qui ont un sarcome d'Ewing localisé peuvent être guéris par cette approche multidisciplinaire. Le traitement est parfois possible même si des métastases existent. La chimiothérapie associée à la résection chirurgicale en bloc lorsqu'elle est possible, donne souvent un meilleur résultat à long terme que la chimiothérapie associée à la radiothérapie.
Fibrosarcome et sarcome pléomorphe indifférencié (anciennement appelé histiocytome fibreux malin de l'os)
Les fibrosarcomes et les sarcomes pléomorphes indifférenciés présentent des caractéristiques similaires à celles des ostéosarcomes mais produisent des cellules tumorales fibreuses (plutôt que des cellules tumorales osseuses); ils affectent la même classe d'âge, et posent des problèmes similaires.
Le traitement et les résultats pour les lésions de haut grade sont similaires à ceux de l'ostéosarcome.
Lymphome de l'os
Le lymphome de l'os (anciennement connu sous le nom de réticulosarcome) affecte les adultes, le plus souvent entre 40 et 60 ans. Il s'agit habituellement d'un lymphome diffus à grandes cellules B. Il peut toucher n'importe quel os. La tumeur est composée de petites cellules rondes, souvent avec un mélange de cellules réticulaires, de lymphoblastes et de lymphocytes. Elle peut se développer comme une tumeur osseuse primitive isolée, en association avec des tumeurs similaires dans d'autres tissus, ou comme une métastase d'une maladie lymphomateuse connue des tissus mous. La douleur et le gonflement sont les symptômes habituels du lymphome de l'os. Les fractures pathologiques sont fréquentes.

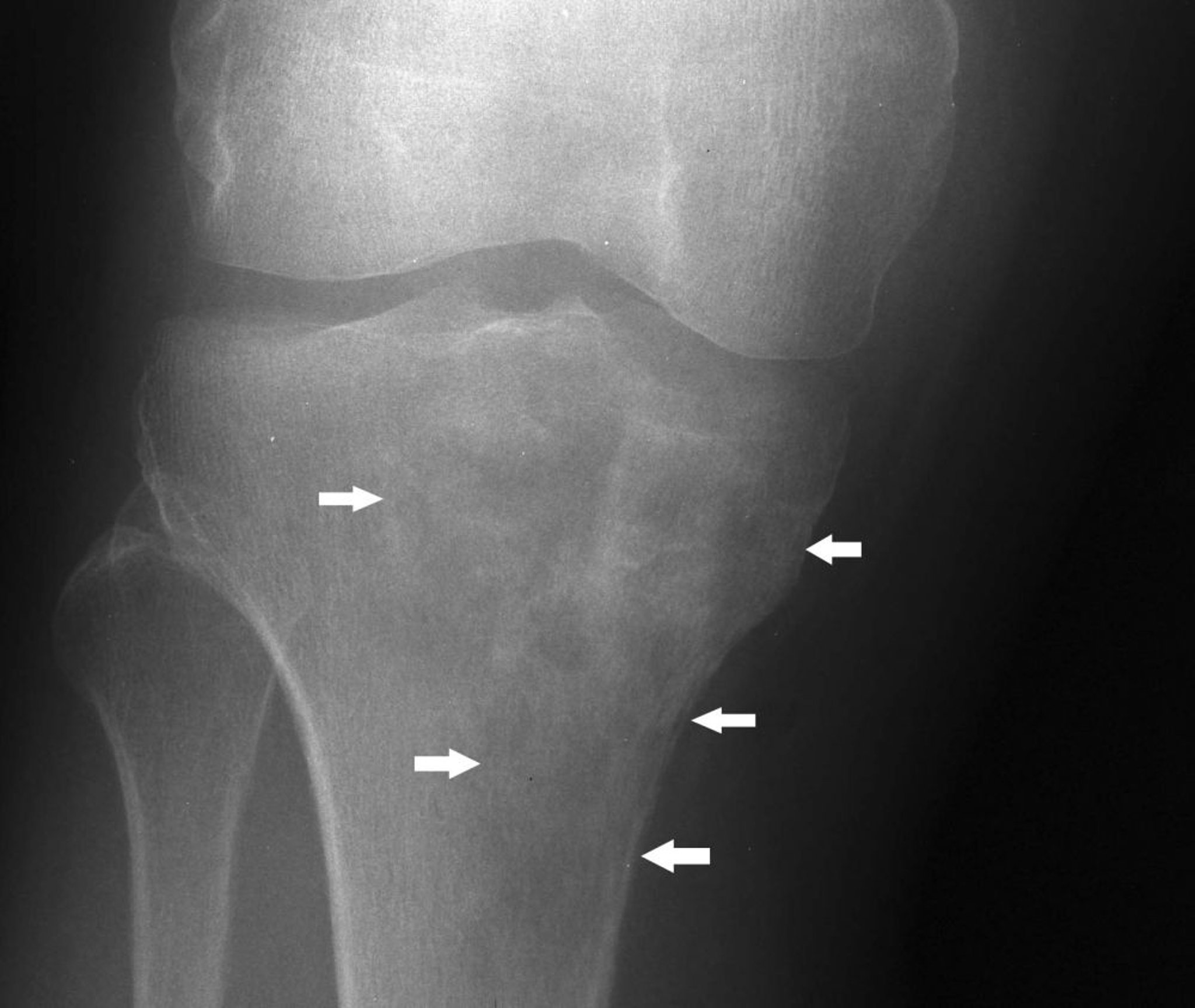
Image courtesy of Michael J. Joyce, MD, et Hakan Ilaslan, MD.
L'imagerie révèle la destruction osseuse, qui peut revêtir un aspect marbré ou tacheté, voire infiltratif et perméatif, souvent avec une volumineuse masse de tissus mous perceptible cliniquement et visible à la rx. A un stade avancé de la maladie, tout le contour de l'os peut avoir disparu. La biopsie est également faite.
Dans le lymphome primitif de l'os isolé, le taux de survie à 5 ans est ≥ 50%.
Les lymphomes osseux sont généralement traités par chimiothérapie systémique. La radiothérapie peut être utilisée comme adjuvant dans certains cas. La consolidation préventive des os longs est souvent nécessaire pour éviter une fracture pathologique. L'amputation n'est que rarement indiquée, lorsque la fonction est perdue du fait d'une fracture pathologique ou de l'extension dans les tissus mous qui ne peut être traitée autrement.
Tumeur maligne à cellules géantes
La tumeur maligne à cellules géantes, qui est rare, est généralement située à l'extrémité d'un os long.
La rx révèle des aspects classiques de destruction maligne (principalement la lyse osseuse, la destruction corticale, l'extension dans les tissus mous, et la fracture pathologique). Une tumeur maligne à cellules géantes qui se développe dans une tumeur à cellules géantes primitivement bénigne est typiquement radiorésistante. L'IRM et la biopsie sont effectuées.
Le traitement des tumeurs malignes à cellules géantes est similaire à celui de l'ostéosarcome, mais le taux de guérison est faible.







