


Le rétinoblastome est un cancer de la rétine, la zone sensible à la lumière située à la partie postérieure de l’œil.
Les rétinoblastomes peuvent être causés par une mutation génétique.
L’enfant peut présenter une pupille blanche ou un strabisme ou parfois des troubles de la vision.
Les médecins peuvent souvent diagnostiquer le rétinoblastome en examinant l’œil à l’aide d’un instrument spécial sous anesthésie.
Le traitement peut comprendre une intervention chirurgicale, une chimiothérapie ou parfois une radiothérapie.
(Voir aussi Présentation des cancers de l’enfant.)
Les rétinoblastomes représentent environ 2 % des cancers de l’enfant et surviennent presque toujours chez les enfants de moins de 2 ans. Les rétinoblastomes ne surviennent que chez les enfants.
Ce cancer est causé par la mutation de certains gènes qui contrôlent le développement de l’œil. Parfois, la mutation est héritée (transmise) d’un parent. D’autres fois, elle survient spontanément (non héritée) très tôt dans le développement de l’embryon.
Lorsque la mutation est héréditaire, les enfants atteints peuvent transmettre la mutation à leurs enfants. La probabilité de transmission de la mutation est de 50 % si un parent présente la mutation. Si la mutation est transmise, la plupart des enfants porteurs de la mutation développeront un rétinoblastome. Le rétinoblastome est héréditaire chez tous les enfants qui développent un cancer des deux yeux, et chez 15 % des enfants qui présentent un cancer d’un seul œil.
D’autres fois, la mutation survient plus tard au cours du développement embryonnaire, uniquement dans les cellules embryonnaires de l’œil. Dans de tels cas, la mutation n’est pas héritée et ne peut pas être transmise aux enfants.
Vue de la rétine

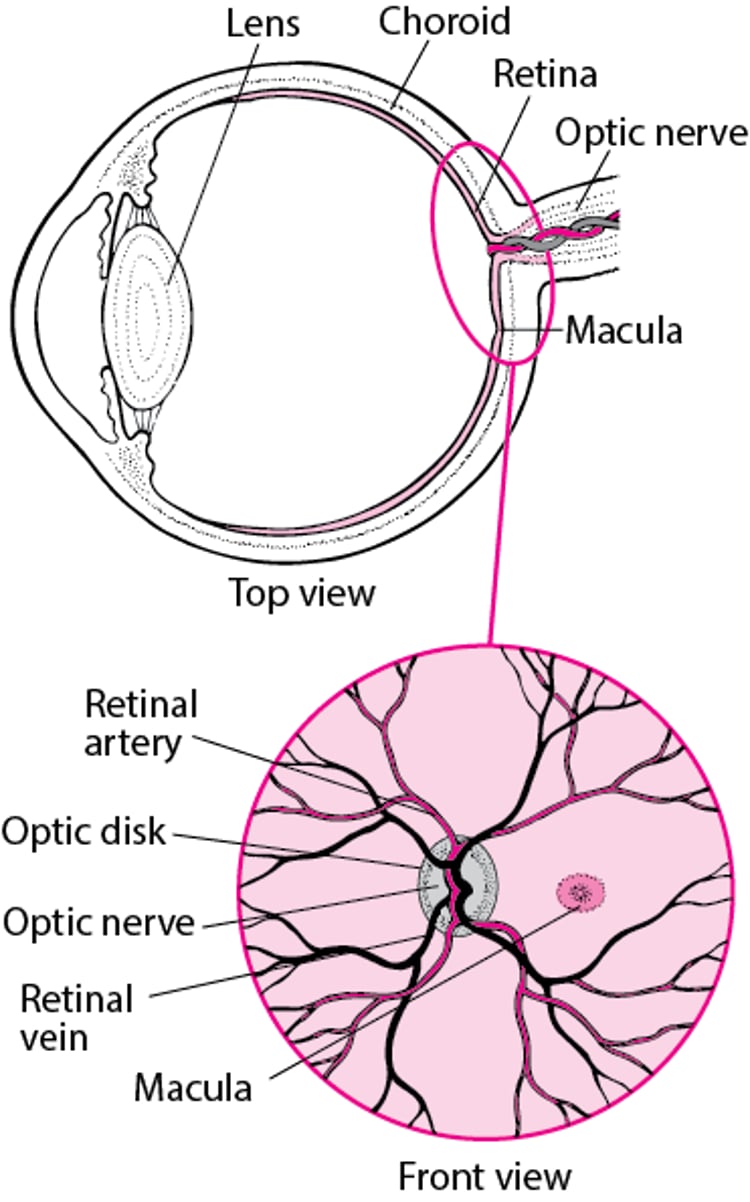
Le rétinoblastome ne se propage en général pas au-delà de l’œil, mais il peut parfois se propager au cerveau par le nerf optique (nerf qui conduit les stimuli de l’œil au cerveau). Il se propage rarement à d’autres endroits, tels que la moelle osseuse et les os.
Symptômes du rétinoblastome
Les symptômes du rétinoblastome peuvent comprendre une pupille blanche (leucocorie) ou une déviation des yeux (strabisme).

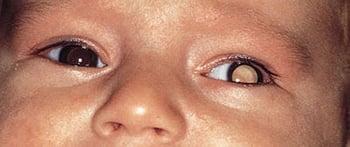

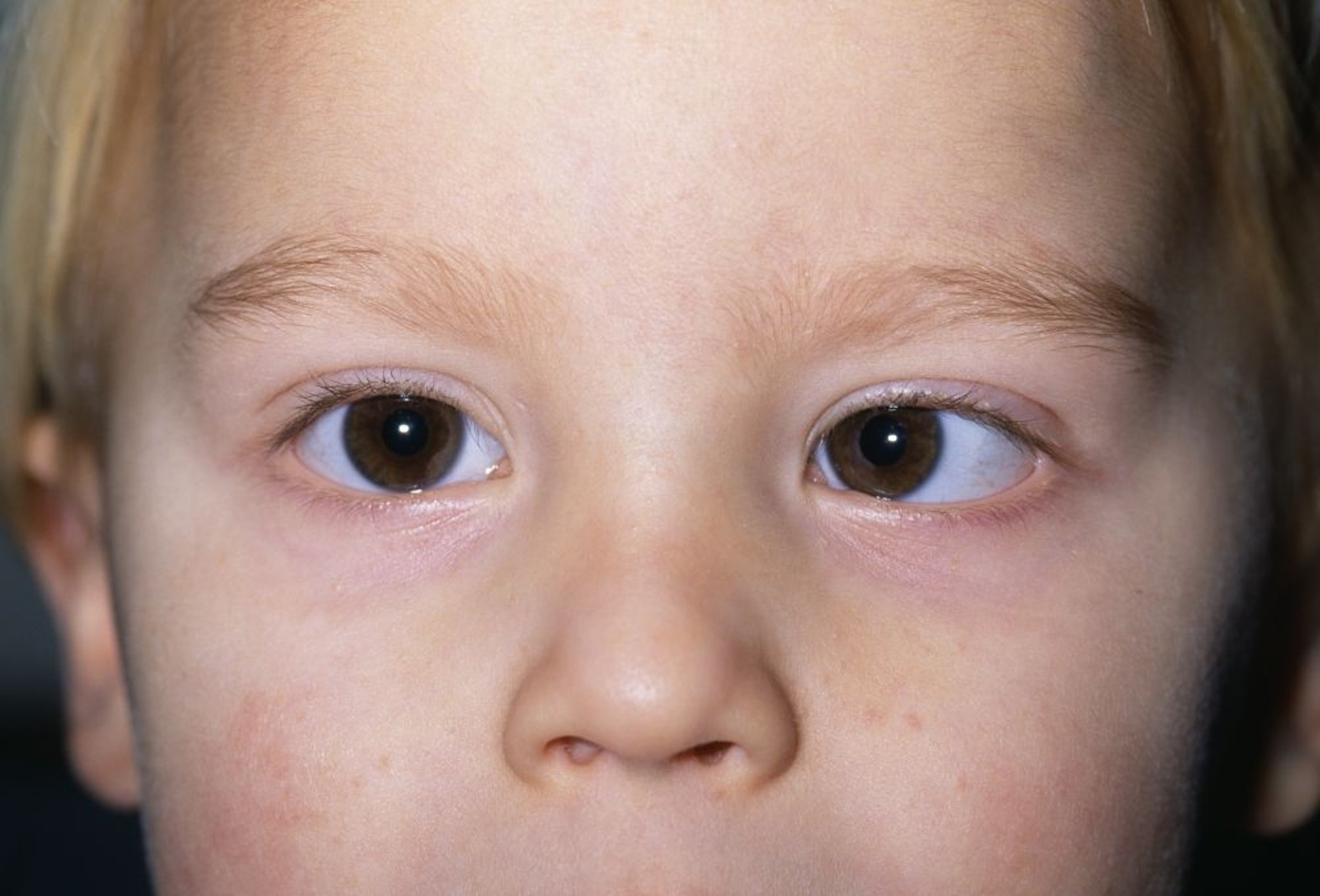
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
Les rétinoblastomes de grande taille peuvent affecter la vision mais tendent à entraîner peu d’autres symptômes.
Si le cancer s’est propagé, les symptômes peuvent inclure des céphalées, une perte de l’appétit ou des vomissements.
Diagnostic du rétinoblastome
Examen de l’œil à l’aide d’un instrument spécial sous anesthésie
Échographie, tomodensitométrie (TDM) ou imagerie par résonance magnétique (IRM) et parfois tomographie à cohérence optique
Parfois scintigraphie osseuse, examen de la moelle osseuse et rachicentèse
Si un médecin suspecte un rétinoblastome, les deux yeux de l’enfant sont examinés sous anesthésie générale (pour que l’enfant ne soit pas conscient). Une lumière et un objectif spécial sont utilisés (ophtalmoscopie indirecte) pour examiner la rétine à travers le cristallin et l’iris. Une anesthésie générale est nécessaire car les petits enfants ne sont pas en mesure de coopérer pendant ce long examen détaillé requis pour le diagnostic du rétinoblastome.
Le cancer peut également être identifié en réalisant une échographie des yeux, une TDM ou une IRM des yeux. Ces examens peuvent également révéler l’existence de métastases cérébrales. La tomographie à cohérence optique est un autre examen d’imagerie qui est parfois réalisé.
Les médecins peuvent également pratiquer une rachicentèse (ponction lombaire) pour rechercher des cellules cancéreuses dans un échantillon de liquide céphalorachidien. L’identification de cellules cancéreuses dans ce liquide constitue une autre preuve que le cancer s’est propagé au cerveau.
Le cancer pouvant se propager jusqu’aux os ou à la moelle osseuse, une scintigraphie osseuse peut être réalisée et un échantillon de moelle osseuse peut être prélevé pour être analysé.
Les enfants souffrant d’un rétinoblastome doivent consulter un généticien et subir des analyses génétiques. Le spécialiste peut alors indiquer aux parents si d’autres membres de la famille sont exposés à un risque et s’il faut réaliser d’autres examens. Habituellement, si l’enfant a un gène de rétinoblastome héréditaire, ses parents et frères et sœurs doivent également subir des examens à la recherche de la mutation génétique. Les frères et sœurs porteurs de la mutation génétique doivent subir des examens oculaires à la recherche d’un rétinoblastome dès la naissance, puis tous les 4 mois jusqu’à l’âge de 4 ans.
Si les analyses génétiques ne sont pas disponibles, tous les enfants ayant un parent, un frère ou une sœur atteints d’un rétinoblastome doivent subir les mêmes examens ophtalmologiques réguliers. Les membres adultes de la famille d’un enfant souffrant d’un rétinoblastome doivent aussi subir un examen ophtalmologique. Même si les adultes ne développeront pas de rétinoblastome, le gène à l’origine de cette pathologie peut également provoquer une tumeur de l’œil non cancéreuse (bénigne) appelée rétinocytome.
Traitement du rétinoblastome
Ablation chirurgicale de l’œil
Chimiothérapie
Radiothérapie, lasers et cryothérapie
(Voir aussi Principes du traitement du cancer et Chirurgie pour le cancer.)
En cas d’atteinte d’un seul œil, et si celui-ci ne possède que peu ou plus d’acuité visuelle, on procède à l’ablation chirurgicale de la totalité du globe oculaire avec une partie du nerf optique.
Lorsque le cancer affecte les deux yeux, les médecins essaient de préserver un certain degré d’acuité visuelle en traitant le cancer sans retirer les deux globes oculaires, bien qu’ils retirent parfois l’œil le plus sévèrement atteint. Les options thérapeutiques incluent les médicaments de chimiothérapie injectés directement dans l’artère principale qui apporte le sang à l’œil (ce que l’on appelle chimiothérapie intra-artérielle), la radiothérapie, les lasers, le gel (cryothérapie), ou pour les cancers de très petite taille, des patchs contenant des matières radioactives (curiethérapie).
Des associations de médicaments de chimiothérapie administrées par voie orale ou intraveineuse (tels que le carboplatine, l’étoposide et la vincristine, ou le cyclophosphamide plus la vincristine) peuvent être utilisées pour réduire la taille d’une grosse tumeur dans un œil, réduire la taille des tumeurs présentes dans les deux yeux, traiter le cancer qui s’est propagé au-delà de l’œil ou traiter le cancer qui réapparaît après le traitement initial. La chimiothérapie est utilisée avec d’autres options thérapeutiques, car seule, elle ne peut généralement pas guérir ce cancer.
La radiothérapie sur l’œil a des effets secondaires significatifs, tels qu’une cataracte, une baisse de l’acuité visuelle, une sécheresse chronique des yeux et une perte des tissus autour de l’œil traité. Les os de la face peuvent ne pas se développer normalement, provoquant des déformations du visage. En outre, le risque de développer un second cancer augmente dans la zone d’administration de la radiothérapie.
Après le traitement, un médecin spécialisé dans le traitement des cancers infantiles (oncologue pédiatrique) et un ophtalmologiste doivent continuer à surveiller l’enfant en raison du risque de développement d’un second cancer.
Pronostic du rétinoblastome
Grâce au traitement, un rétinoblastome qui ne s’est pas propagé au-delà de la rétine guérit dans plus de 90 % des cas. Le pronostic est défavorable pour les enfants dont le cancer s’est propagé.
En l’absence de traitement, la plupart des enfants qui souffrent d’un rétinoblastome décèdent dans les 2 ans.
Les enfants souffrant d’un rétinoblastome de type héréditaire sont exposés à un risque accru de développer un second cancer, tel que le sarcome des tissus mous, le mélanome ou l’ostéosarcome. Environ la moitié des seconds cancers se développent au site d’administration de la radiothérapie. Dans environ 70 % des cas, le second cancer se développe dans les 30 ans suivant le rétinoblastome.
Informations supplémentaires
Les ressources en anglais suivantes pourraient vous être utiles. Veuillez noter que LE MANUEL n’est pas responsable du contenu de cette ressource.
American Cancer Society (Société américaine du cancer) : If Your Child Is Diagnosed with Cancer (Si votre enfant reçoit un diagnostic de cancer) : ressource pour les parents et les proches d’un enfant atteint d’un cancer qui fournit des informations sur la manière de faire face à certains problèmes et questions qui surgissent juste après le diagnostic.




