

Charcot-Marie-Tooth disease is a hereditary neuropathy in which the muscles of the lower legs become weak and waste away (atrophy).
Charcot-Marie-Tooth disease affects nerves that control muscle movement and those that carry sensory information to the brain.
Weakness begins in the lower legs and gradually moves up the limbs, and people lose the ability to sense vibration, pain, and temperature.
Electromyography and nerve conduction studies are done to confirm the diagnosis.
No treatment can stop the progression of the disease, but the use of braces and physical and occupational therapy may help people function better.
(See also Overview of the Peripheral Nervous System.)
Charcot-Marie-Tooth disease is the most common hereditary neuropathy, affecting about 1 of 2,500 people. It may begin during childhood or later in life.
Charcot-Marie-Tooth disease is a sensory and motor neuropathy. That is, it affects motor nerves (which control muscle movement) and sensory nerves (which carry sensory information to the brain).
There are several types of Charcot-Marie-Tooth disease. But typically, the disease is categorized based on what type of damage it causes, as follows:
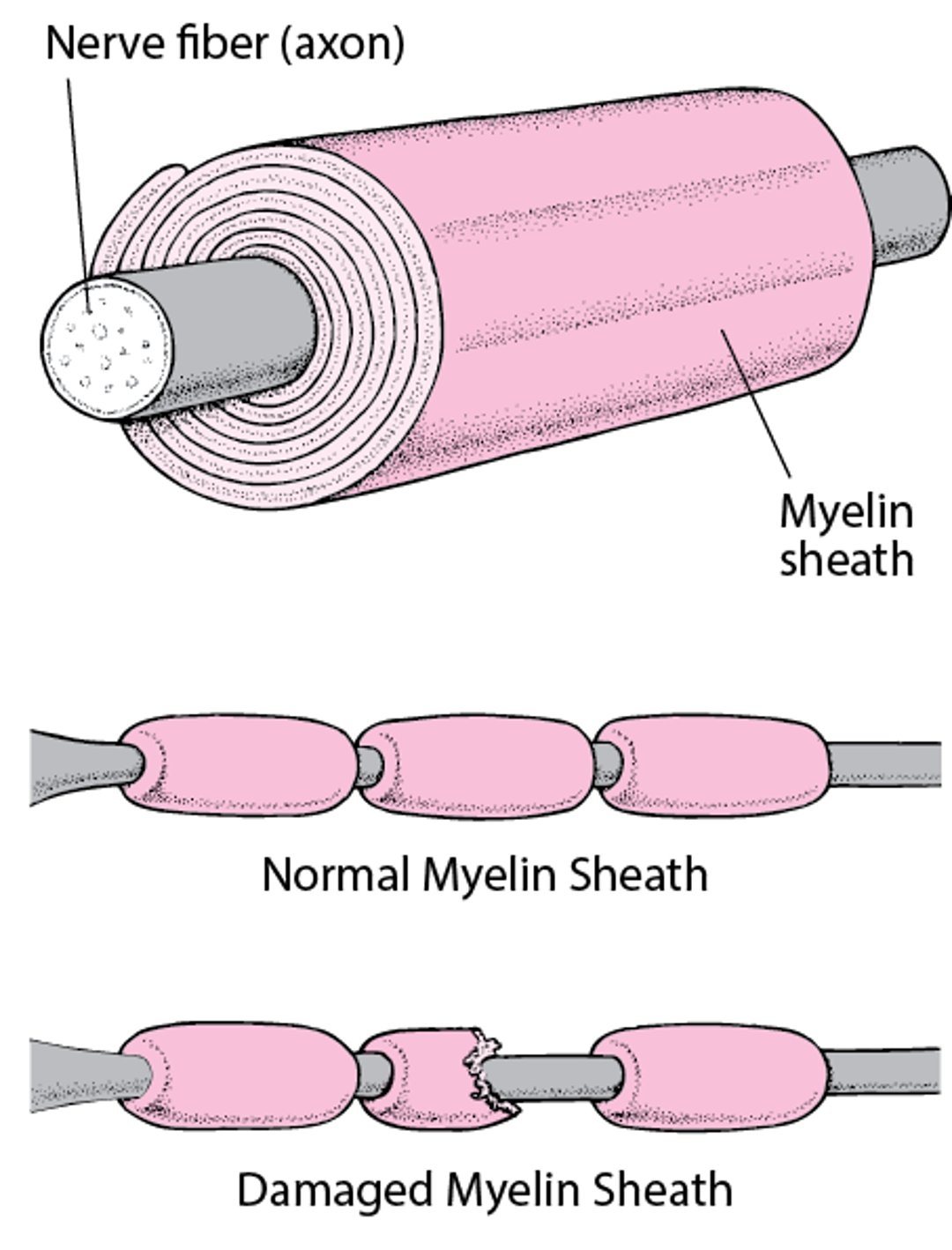
Demyelination (damage to the myelin sheath): The axons (the part of the nerve that sends messages) remain alive, but the myelin sheath surrounding them is damaged or destroyed (demyelinated). (The myelin sheath functions somewhat like insulation around electrical wires, enabling nerve impulses to travel quickly.)
Damage to the axon: The axon (the part of the nerve that sends messages) is mainly affected. Sometimes the axons die.
Insulating a Nerve Fiber
Most nerve fibers inside and outside the brain are wrapped with many layers of tissue composed of a fat (lipoprotein) called myelin. These layers form the myelin sheath. Much like the insulation around an electrical wire, the myelin sheath enables nerve signals (electrical impulses) to be conducted along the nerve fiber with speed and accuracy. When the myelin sheath is damaged (called demyelination), nerves do not conduct electrical impulses normally. |

Most types of Charcot-Marie-Tooth disease are usually inherited as an autosomal (not sex-linked) dominant trait. That is, only one gene from one parent is required for the disease to develop. However, some forms can be inherited as a recessive trait (requiring two genes, one from each parent) or sex (X)-linked. In sex-linked inheritance, the gene is on the X chromosome. This chromosome determines whether a person is male or female. Males have one X chromosome (from their mother) and one Y chromosome (from their father). Females have two X chromosomes (one from their mother and one comes from their father). If a male inherits an X chromosome with the abnormal gene, he develops the disease. If a female inherits one abnormal X chromosome, she is unlikely to develop the disease because she has also inherited one normal X chromosome.
Dejerine Sottas disease (hypertrophic interstitial neuropathy) is a rare hereditary sensory and motor neuropathy. It causes symptoms similar to those of Charcot-Marie-Tooth disease. However, weakness worsens much more quickly. Sensation and reflexes are also lost. It begins during childhood. It can be inherited as an autosomal (not sex-linked) dominant or recessive trait. That is, for the dominant form, only one gene from one parent for the disease is required for the disease to develop, and for the recessive form, two genes, one from each parent, are required.
Symptoms of Charcot-Marie-Tooth Disease
Symptoms of Charcot-Marie-Tooth disease vary depending on the type.
In one type, symptoms may begin in middle childhood or adolescence. Weakness begins in the lower legs. It causes an inability to flex the ankle to lift the front part of the foot (footdrop) and wasting away of the calf muscles (stork leg deformity). Later, hand muscles begin to waste away. The hands and feet become unable to sense position, vibration, pain, and temperature, and this loss of sensation gradually moves up the limbs.
In milder types of the disease, high arches and hammer toes may be the only symptoms. In one type, males have severe symptoms, and females have mild symptoms or may be unaffected.
The disease progresses slowly and does not affect life span.
Diagnosis of Charcot-Marie-Tooth Disease
A doctor's evaluation
Electromyography and nerve conduction studies
Asking about the following can help doctors diagnose Charcot-Marie-Tooth disease:
Which areas of the body are weak
When the disease began
Whether family members have similar symptoms
They also check whether people have foot deformities (high arches and hammer toes). This information helps doctors identify the different types of Charcot-Marie-Tooth disease and distinguish them from other causes of neuropathy.
Electromyography and nerve conduction studies are done to confirm the diagnosis.
Genetic testing and counseling for Charcot-Marie-Tooth disease are available.
Treatment of Charcot-Marie-Tooth Disease
Braces for footdrop
Sometimes physical and occupational therapy
No treatment can stop the progression of Charcot-Marie-Tooth disease.
Wearing braces helps correct footdrop, and sometimes orthopedic surgery is needed to stabilize the foot.
Physical therapy (to strengthen muscles) and occupational therapy may be helpful. Vocational counseling may help people maintain vocational skills even though the disorder is progressing.





