


Amyloidosis is a rare disease in which abnormally folded proteins form into collections called amyloid fibrils that accumulate in various tissues and organs, sometimes leading to organ dysfunction, organ failure, and death.
The symptoms and severity of amyloidosis depend on which vital organs are affected.
Diagnosis is made by obtaining a sample of tissue (biopsy) and examining it under a microscope.
There are many forms of amyloidosis, and other testing is required to identify the form and its cause.
Treatment depends upon the type of amyloidosis.
Causes of Amyloidosis
Proteins are long chains of molecules that fold up into a certain shape. The exact shape is critical to how each protein functions. All types of amyloidosis involve a protein that folds abnormally. The abnormally folded proteins clump together and accumulate in various body tissues as amyloid deposits. These deposits are made up of amyloid fibrils, which contain abnormal protein fibers that cannot be easily broken down by the body. These deposits interfere with normal function of the organ where they are located. All of these proteins are produced within the body and do not come from a person's diet. Some amyloid proteins are mutated versions of normal proteins. Others are normal proteins that simply have a tendency to fold abnormally. Some of the proteins are produced in various other diseases (such as tuberculosis or rheumatoid arthritis).
Forms of Amyloidosis
Amyloid deposits may be
Systemic: widespread throughout the body
Localized: affecting only one organ or tissue
The severity of the disease depends on which organs are affected by the amyloid deposits.
Systemic amyloidosis
Systemic amyloidosis can be classified into four major groups:
AL (primary) amyloidosis (light chain amyloidosis) occurs with abnormalities of plasma cells (a type of immune cell that makes antibodies) that cause them to produce excessive amounts of abnormal antibody proteins called light chains. Some people with AL amyloidosis also have cancer of the plasma cells (multiple myeloma). Only about 10 to 20% of people with multiple myeloma develop AL amyloidosis. Typical sites of amyloid buildup in AL amyloidosis are the skin, heart, kidneys, nerves, tongue, intestine, liver, spleen, and blood vessels.
AA (secondary) amyloidosis may develop in response to various diseases that cause persistent infection or inflammation (such as tuberculosis, rheumatoid arthritis, and familial Mediterranean fever) and certain types of cancer. AA amyloidosis most often leads to kidney disease, although other organs may also be affected.
AF (familial) amyloidosis is a collection of rare inherited diseases that causes symptoms in adulthood. The amyloid-producing defect occurs because of inherited mutations in specific proteins in the blood. These mutated proteins form amyloid fibrils that typically target the kidneys, nerves, or heart. Mutated transthyretin, a protein produced by the liver, is the most frequent cause of familial amyloidosis.
ATTRwt (wild type transthyretin) amyloidosis (previously called senile systemic amyloidosis) usually affects the heart. It is much more common in men than in women. ATTRwt amyloidosis is caused by abnormal folding of normal (wild type, not mutated) transthyretin protein. It is not known what causes amyloid to build up in the heart.
Localized amyloidosis
Localized amyloidosis occurs when amyloid is deposited in specific organs or tissues. For example, amyloid accumulates in the brain of people with Alzheimer disease and is thought to play a role in causing Alzheimer disease. Localized amyloid deposits may also form in the skin, digestive tract, airways, or bladder.
Symptoms of Amyloidosis
The accumulation of large amounts of amyloid deposits can disturb the normal functioning of many organs. Some people have few symptoms, whereas others develop severe, life-threatening disease. Common symptoms of amyloidosis are fatigue and weight loss. Other symptoms of amyloidosis depend on where the amyloid deposits build up.
When the heart is affected, people may have abnormal heart rhythms or heart failure, which may cause shortness of breath, weakness, or fainting.
When nerves are affected, people may have tingling or numbness in the fingers or toes or dizziness when standing up. When the kidneys are affected, people may have swelling (edema) of the feet and legs and sometimes the abdomen.

© Springer Science+Business Media
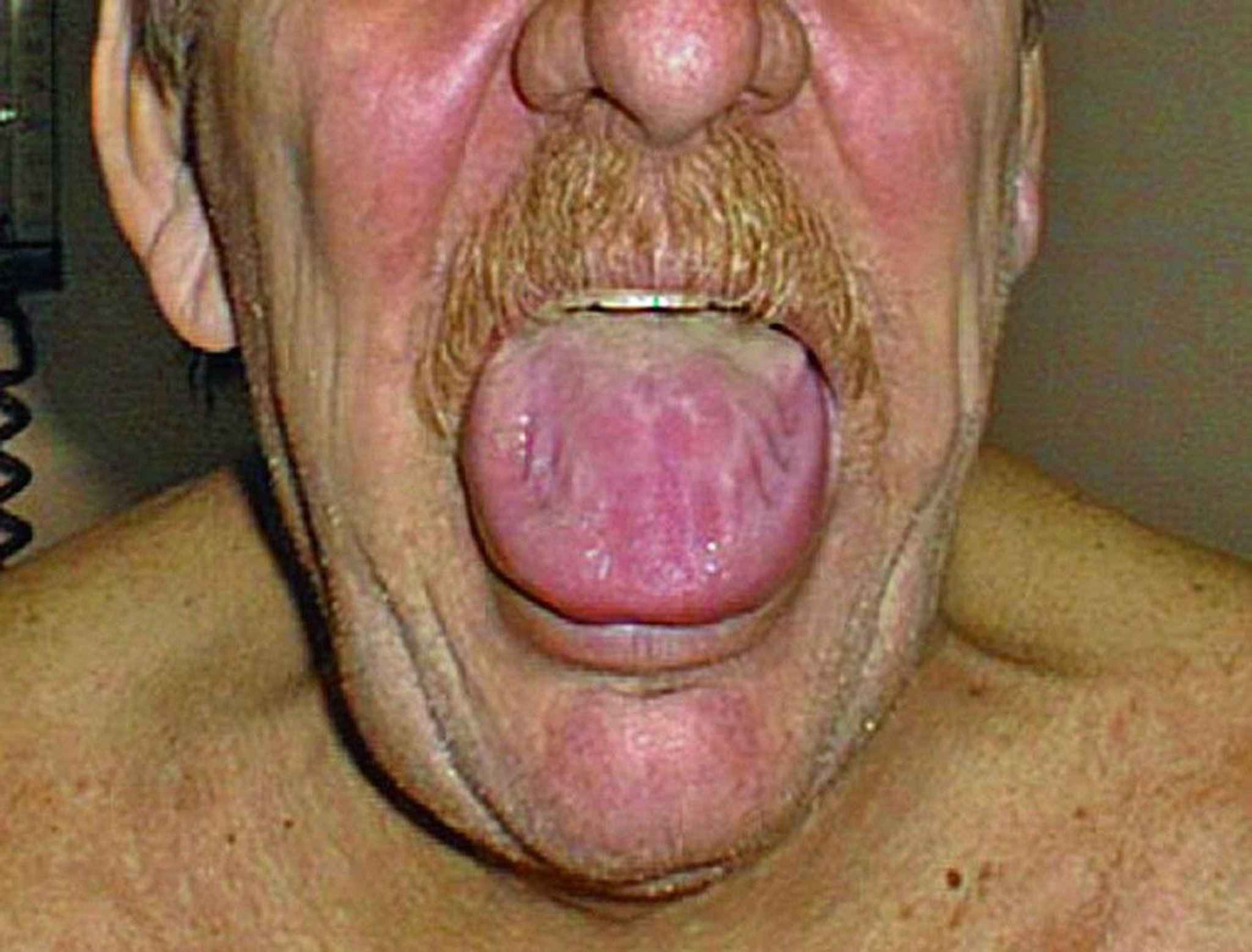
When the skin is involved, easy bruising is common, which can sometimes occur around the eyes. The tongue sometimes enlarges (macroglossia).
Diagnosis of Amyloidosis
Biopsy
Amyloidosis is sometimes difficult for doctors to recognize because it causes so many different problems, and many of these problems are also caused by other diseases. Doctors suspect amyloidosis when people have symptoms involving multiple organs of the body, or if they have unexplained heart, kidney, or liver failure. Macroglossia is not a common symptom, but when it occurs it is suggestive of amyloidosis. When several family members have heart or nerve problems, it may suggest AF amyloidosis. Unexplained heart symptoms in older men is suggestive of ATTRwt amyloidosis.
The diagnosis is generally made by testing a small amount of abdominal fat obtained through a needle inserted in the belly (fat pad biopsy). Alternatively, doctors can do a biopsy by taking a sample of tissue from a part of the body that has been affected by the amyloidosis, such as the heart, kidneys, or liver, and examining it under a microscope with the use of special stains.
After doctors determine the person has amyloidosis, they do other tests to determine the type of amyloidosis and to identify any disorders that may be the cause of the amyloidosis. They also do tests to see what organs have been affected.
Treatment of Amyloidosis
In AL amyloidosis, chemotherapy
In AA amyloidosis, treatment of the underlying disease
In amyloidosis caused by transthyretin protein deposits, medications that stabilize transthyretin
Sometimes, organ transplantation
Treatment to decrease or control symptoms and complications of amyloidosis can improve quality of life for people with all forms of amyloidosis. Specific treatments to slow or stop amyloid formation can help in certain forms of amyloidosis.
For people with AL amyloidosis,stem cell transplant, can arrest the disease in the bone marrow, and prevent progression of the amyloid deposits. Radiation therapy can be used in people whose AL amyloidosis is present in only one area (localized disease).
For AA (secondary) amyloidosis,
For amyloidosis caused by transthyretin protein deposits,
Organ transplants (for example, a kidney or the heart) have extended the lives of some people with organ failure due to amyloidosis.
In familial transthyretin amyloidosis,liver transplantation may be used. Liver transplantation can slow progression of the disease because the liver is where the mutant protein is produced. Interestingly, because there is a shortage of donor organs, the liver removed from a person with familial transthyretin amyloidosis is sometimes transplanted into people with a fatal liver disease such as cirrhosis or liver cancer. Such a "domino transplant" is possible because a liver from a person with familial transthyretin amyloidosis is otherwise a normally functioning liver. Although people who receive a liver from a person with familial transthyretin amyloidosis may eventually develop amyloidosis themselves, the transplant can save their lives in the short term.
Prognosis for Amyloidosis
The prognosis depends on the type of amyloidosis and the organ system that is affected. Involvement of the heart is the most dangerous form and can have a bleak prognosis.




