


La malattia di Huntington è una malattia ereditaria che inizia con spasmi o scatti occasionali involontari e in seguito progredisce a movimenti involontari più pronunciati (corea e atetosi), deterioramento mentale e decesso.
Nella malattia di Huntington si assiste alla degenerazione delle parti del cervello che rendono fluidi e coordinano i movimenti.
Questi ultimi diventano convulsi e scoordinati e la funzione mentale, che include l’autocontrollo e la memoria, si deteriora.
Il medico basa la diagnosi sui sintomi, sull’anamnesi familiare, sugli esami di diagnostica per immagini del cervello e sui test genetici.
I farmaci possono aiutare a calmare i sintomi, ma la malattia è progressiva e porta alla morte.
(Vedere anche Panoramica sui disturbi del movimento.)
La malattia di Huntington colpisce tra 1 e 10 persone su 100.000. Il numero di soggetti colpiti varia a seconda della parte del mondo in cui vivono. Interessa entrambi i sessi allo stesso modo.
Il gene della malattia di Huntington è dominante. Quindi, se si ha una sola copia del gene anomalo, ereditato da un genitore, questa è sufficiente per causare la malattia. Di conseguenza, i figli di una persona con la malattia di Huntington hanno il 50% di probabilità di svilupparla.
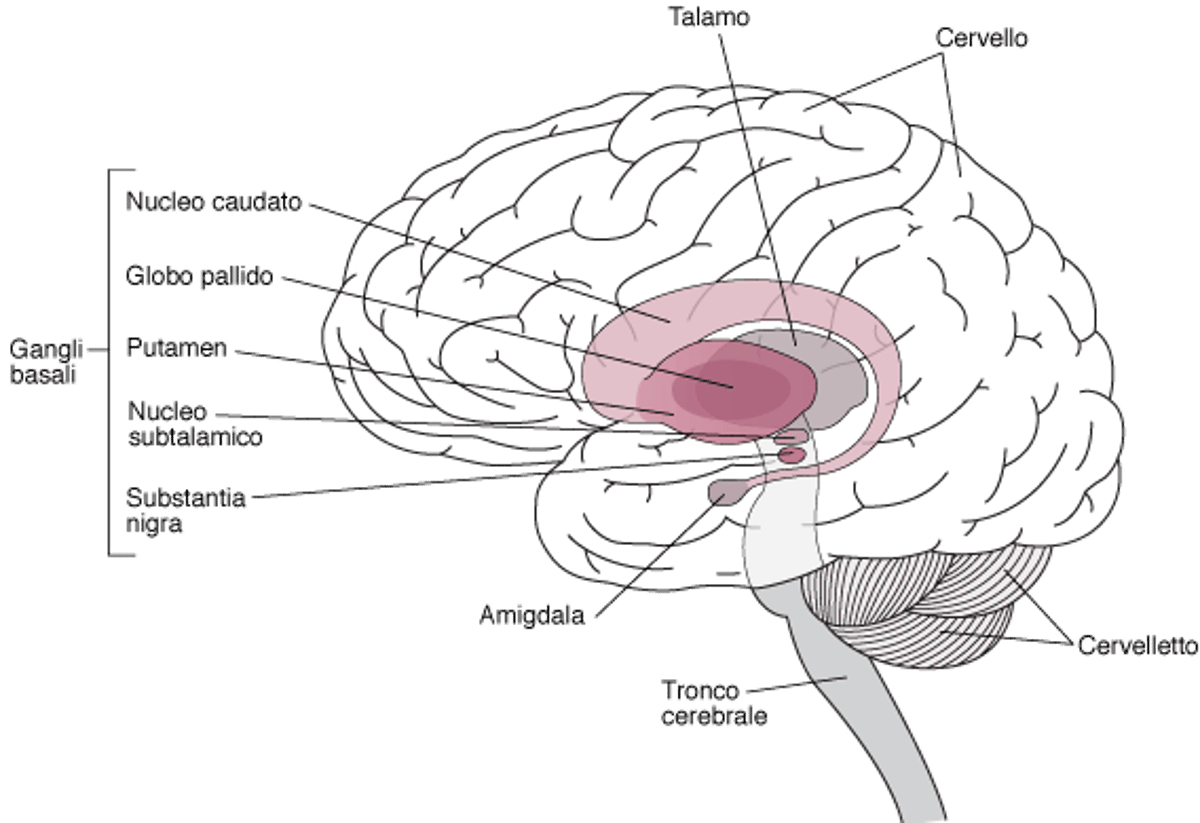
La malattia di Huntington è causata da una degenerazione graduale di parti dei gangli basali dette nucleo caudato e putamen. I gangli basali sono gruppi di cellule nervose situate alla base del cervello, in profondità all’interno dell’encefalo. I gangli basali coordinano e rendono fluidi i movimenti.
Localizzazione dei gangli basali
I gangli basali sono aggregati di cellule nervose situati in profondità nel cervello. Tra questi vi sono i seguenti:
I gangli basali aiutano ad avviare ed effettuare movimenti volontari fluidi, a sopprimere i movimenti involontari e a coordinare i cambiamenti di postura. |

Sintomi della malattia di Huntington
I sintomi della malattia di Huntington si sviluppano solitamente in modo impercettibile, iniziano fra i 35 e i 40 anni di età, ma a volte prima dell’età adulta.
Negli stati iniziali della malattia di Huntington, il viso, il tronco e gli arti possono muoversi involontariamente e rapidamente. All’inizio, le persone possono camuffare i movimenti involontari anormali con movimenti volontari, in modo da non far notare quelli anormali. Tuttavia, con il tempo questi diventano più ovvi.
I muscoli possono contrarsi brevemente e rapidamente, provocando improvvisi spasmi delle braccia o di altre parti del corpo, talvolta varie volte di seguito.
Le persone possono camminare in modo ritmato o esageratamente spavaldo, come burattini. Possono fare smorfie, muovere gli arti a scatti e ammiccare più spesso. I movimenti diventano scoordinati e lenti. Alla fine viene colpito tutto il corpo, rendendo estremamente difficile camminare, stare seduti, mangiare, parlare, deglutire e vestirsi.
Si manifestano frequentemente cambiamenti mentali prima o quando si sviluppano i movimenti anormali. Questi cambiamenti sono inizialmente impercettibili. Le persone possono diventare gradualmente irritabili, eccitabili e agitate. Possono perdere interesse nelle attività normali. Possono non essere in grado di controllare gli impulsi, perdendo la pazienza, avendo attacchi di sconforto, oppure diventando promiscue.
Con il progredire della malattia di Huntington, possono comportarsi in modo irresponsabile e spesso vagare senza meta. Con gli anni, perdono la memoria e la capacità di pensare in modo razionale. Possono diventare gravemente depresse e tentare il suicidio. Possono anche diventare ansiosi o sviluppare un disturbo ossessivo-compulsivo.
Nello stadio avanzato, la demenza è grave e le persone sono confinate a letto. È necessaria un’assistenza a tempo pieno o cure infermieristiche a domicilio. La morte sopravviene 13-15 anni dopo l’insorgenza dei sintomi.
Diagnosi della malattia di Huntington
Valutazione del medico, confermata da test genetici.
Tomografia computerizzata o risonanza magnetica per immagini
La malattia di Huntington può essere difficile da riconoscere nella sua fase iniziale, perché i sintomi sono impercettibili. Può essere sospettata sulla base dei sintomi e dell’anamnesi familiare. Si deve comunicare al medico se ci sono parenti con problemi mentali o ai quali sono stati diagnosticati disturbi neurologici (come la malattia di Parkinson) o disturbi psichiatrici (come la schizofrenia) perché potrebbero aver avuto una malattia di Huntington non diagnosticata.
Vengono effettuate una tomografia computerizzata (TC) o una risonanza magnetica per immagini (RMI) per controllare la degenerazione dei gangli basali e delle altre aree del cervello solitamente colpite dalla malattia e per escludere altri disturbi.
Per confermare la diagnosi si eseguono test genetici. I test genetici e la consulenza sono importanti per le persone con un’anamnesi familiare della malattia ma nessun sintomo, perché potrebbero avere figli prima della comparsa dei sintomi. Per queste persone, la consulenza genetica deve precedere il test genetico. Vengono inviate presso dei centri specializzati nella gestione delle complesse questioni etiche e psicologiche implicate.
Trattamento della malattia di Huntington
Antipsicotici e altri farmaci per alleviare i sintomi
Non appena possibile dopo la diagnosi, le persone con la malattia di Huntington dovrebbero redigere direttive anticipate, indicando il tipo di cura medica che preferiscono nella fase terminale.
Non esistono cure per la malattia di Huntington. Tuttavia, alcuni farmaci, inclusi gli antipsicotici (come clorpromazina, aloperidolo, risperidone e olanzapina), possono aiutare a controllare l'agitazione. I farmaci che riducono la quantità di dopamina (come tetrabenazina, deutetrabenazina e l'antipertensivo reserpina) possono aiutare a fermare (sopprimere) i movimenti alterati.
Si possono utilizzare gli antidepressivi per trattare la depressione, se presente.
I medici offrono consulenza genetica e analisi genetiche ai soggetti i cui genitori o fratelli sono affetti dalla malattia di Huntington. La consulenza genetica deve essere offerta prima delle analisi genetiche, perché le conseguenze della malattia di Huntington sono molto gravi. La consulenza è particolarmente importante per le donne in età fertile e per gli uomini che intendono procreare.
Ulteriori informazioni
Di seguito si riporta una risorsa in lingua inglese che può essere utile. Si prega di notare che il MANUALE non è responsabile del contenuto di questa risorsa.
Genetics Home Reference: Huntington disease: Questo sito Web descrive la malattia di Huntington, i sintomi che provoca e come viene ereditata, e fornisce dei collegamenti relativi alla sua diagnosi e trattamento.