


I disturbi che possono causare la demielinizzazione che non hanno una causa nota sono detti patologie demielinizzanti primarie. La demielinizzazione è la distruzione dei tessuti che avvolgono i nervi, chiamati guaina mielinica.
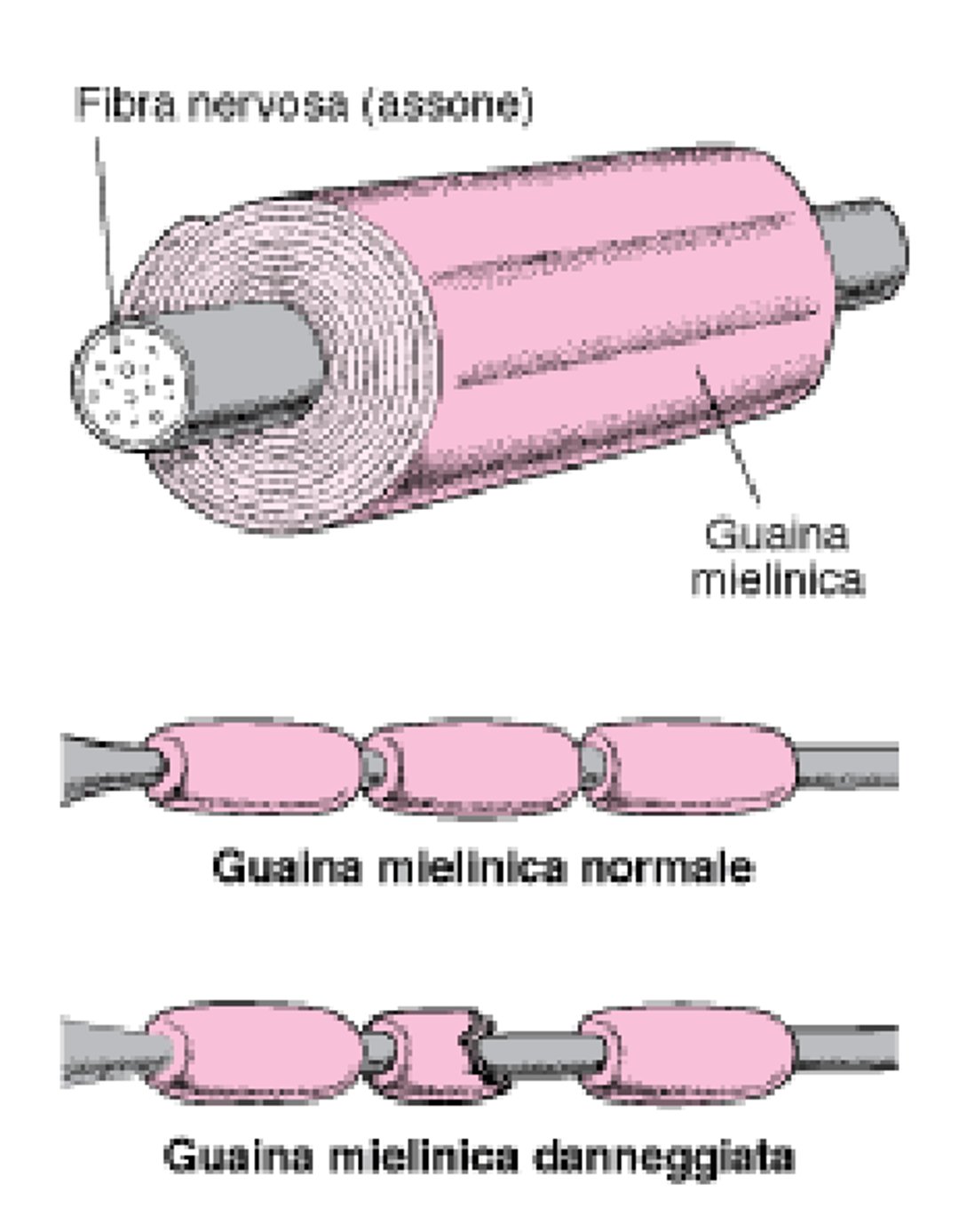
Isolamento di una fibra nervosa
La maggior parte delle fibre nervose all’interno e all’esterno del cervello sono avvolte in molti strati di tessuto composto da un grasso (lipoproteina) chiamato mielina. Questi strati formano la guaina mielinica. Come l’isolamento intorno a un cavo elettrico, la guaina mielinica consente ai segnali nervosi (impulsi elettrici) di essere condotti lungo le fibre nervose in modo veloce e preciso. Quando la guaina mielinica è danneggiata (demielinizzazione), i nervi non conducono gli impulsi elettrici in modo normale. |

Talvolta le patologie demielinizzanti primarie si sviluppano dopo un’infezione virale o una vaccinazione contro un’infezione virale. Una spiegazione probabile è che il virus o un’altra sostanza stimola in qualche modo il sistema immunitario ad attaccare i tessuti stessi dell’organismo (reazione autoimmune). La reazione autoimmune conduce a un’infiammazione, che danneggia la guaina mielinica e le fibre nervose sottostanti.
La sclerosi multipla è la patologia demielinizzante primaria più comune.
Encefalomielite acuta disseminata (ADEM)
Questo raro tipo di infiammazione porta alla demielinizzazione dei nervi nel cervello e nel midollo spinale. L’encefalomielite acuta disseminata è più comune nei bambini che negli adulti.
L’encefalomielite acuta disseminata solitamente si sviluppa dopo un’infezione virale. Si pensa che l’encefalomielite acuta disseminata sia una reazione immune alterata scatenata dal virus. Negli Stati Uniti, questa malattia deriva solitamente da alcuni tipi di influenza, epatite A, epatite B oppure infezione da enterovirus, virus di Epstein-Barr o virus da immunodeficienza umana (HIV). Morbillo, varicella e rosolia erano considerate cause comuni prima che venisse diffusa la vaccinazione nell’infanzia.
Solitamente l’infiammazione si sviluppa 1-3 settimane dopo l’inizio della malattia virale.
Sintomi della ADEM
I sintomi dell’encefalomielite acuta disseminata compaiono rapidamente. Inizialmente si può sviluppare febbre, cefalea, nausea e vomito e sensazione di stanchezza. Se il disturbo è grave, può provocare convulsioni e coma.
Si può perdere la vista in uno o entrambi gli occhi. I muscoli possono indebolirsi e la coordinazione può risultare compromessa, rendendo difficile la deambulazione. Il soggetto può diventare paralizzato. Parti del corpo possono perdere la sensibilità, diventando intorpidite. Può essere colpita la funzione mentale (compresi pensiero, giudizio e apprendimento).
La maggior parte delle persone recupera entro pochi giorni, ed entro 6 mesi la maggior parte ha recuperato completamente o quasi completamente. Altre persone possono rimanere compromesse per il resto della vita. I muscoli possono rimanere deboli e parti del corpo possono rimanere intorpidite. È possibile che le persone non recuperino la vista o la funzione mentale.
Diagnosi della ADEM
Valutazione medica
I medici possono essere in grado di diagnosticare l’encefalomielite acuta disseminata in base ai sintomi e ai risultati di un esame obiettivo. Può essere effettuata una risonanza magnetica per immagini (RMI).
Viene effettuata una puntura lombare (rachicentesi) se si sospetta una meningite o un’infezione del cervello (encefalite). Possono essere effettuati degli esami del sangue per controllare la presenza di altri disturbi che potrebbero causare sintomi simili.
Trattamento della ADEM
Corticosteroidi
Immunoglobulina o plasmaferesi
L’encefalite acuta disseminata può essere trattata con corticosteroidi somministrati per via endovenosa.
Anche le immunoglobuline e la plasmaferesi possono essere efficaci. Questi trattamenti possono essere utilizzati con o senza corticosteroidi. Le immunoglobuline sono costituite da anticorpi ottenuti dal sangue di persone con un sistema immunitario sano. Nella plasmaferesi viene prelevato del sangue, gli anticorpi anomali vengono rimossi e il sangue viene nuovamente trasfuso nella persona.
Adrenoleucodistrofia e adrenomieloneuropatia
L’adrenoleucodistrofia e l’adrenomieloneuropatia sono rari disturbi metabolici ereditari. In questi disturbi, i grassi non vengono scomposti normalmente. Questi grassi si accumulano principalmente nel cervello, nel midollo spinale e nelle ghiandole surrenali. Nel cervello, causano demielinizzazione dei nervi.
L’adrenoleucodistrofia colpisce i bambini di sesso maschile, solitamente di età compresa fra i 4 e gli 8 anni. Una forma più lieve e a sviluppo più lento della malattia può iniziare durante l’adolescenza o la prima età adulta.
L’adrenomieloneuropatia è una forma più lieve. Inizia quando gli uomini hanno tra venti e quarant’anni.
In queste malattie, la demielinizzazione diffusa è spesso accompagnata da disfunzione della ghiandola surrenale. I ragazzi hanno problemi di comportamento, di udito e di vista. Alla fine si manifestano deterioramento mentale, contrazioni muscolari involontarie e scoordinate (spasticità) e cecità. Alcuni ragazzi con adrenoleucodistrofia sono totalmente disabili o muoiono entro 2-3 anni dalla diagnosi. Spesso gli adulti con adrenomieloneuropatia notano inizialmente un problema alle gambe, che diventano deboli e rigide, poi perdono il controllo della vescica o dell’intestino (incontinenza) e/o si sviluppa una disfunzione erettile.
La diagnosi di adrenoleucodistrofia o adrenomieloneuropatia è confermata da test genetici.
Non si conosce alcuna cura per queste malattie. Gli integratori alimentari a base di glicerolo trioleato e glicerolo trierucato (noto anche come olio di Lorenzo) possono aiutare, ma sono necessari ulteriori studi.
Quando viene colpita la ghiandola surrenale (ma non il cervello), il trattamento con ormoni surrenali può salvare la vita. Molti esperti ora raccomandano il trapianto di cellule staminali nelle fasi precoci della malattia prima che si sviluppino sintomi gravi.
Neuropatia ottica ereditaria di Leber
La neuropatia ottica ereditaria di Leber causa una demielinizzazione che porta a cecità parziale.
La neuropatia ottica ereditaria di Leber è più comune tra i pazienti di sesso maschile. Di solito, i sintomi esordiscono a un’età compresa tra i 15 e i 35 anni. Questa malattia viene ereditata dalla madre e i geni difettosi sembrano essere localizzati nel mitocondrio (strutture nelle cellule che forniscono loro energia).
La vista può diventare offuscata in uno o in entrambi gli occhi contemporaneamente. Ma se è colpita la vista di un solo occhio, la vista dell’altro occhio inizia a scomparire entro settimane o mesi. L’acuità visiva e la visione a colori si deteriorano nel tempo.
Alcune persone hanno anche problemi cardiaci o sintomi muscolari (come contrazioni involontarie dei muscoli, debolezza muscolare o spasmi muscolari), che possono assomigliare ai sintomi della sclerosi multipla.
I medici sono spesso in grado di diagnosticare la neuropatia ottica ereditaria di Leber in base ai sintomi e ai risultati dell’esame obiettivo. Gli esami possono identificare alcuni geni anomali responsabili delle malattie. L’elettrocardiogramma viene eseguito per verificare la presenza di problemi di cuore.
Non esiste alcun trattamento stabilito per la neuropatia ottica ereditaria di Leber. Tuttavia, alcune evidenze suggeriscono che i farmaci idebenone e ubichinone possono migliorare la vista in soggetti con neuropatia ottica ereditaria di Leber. La terapia genica è in fase di studio. Essa prevede l’iniezione del gene normale nell’occhio.
Può essere utile limitare il consumo di alcol e non usare prodotti a base di tabacco. L’alcol e il tabacco possono influire sui mitocondri, in cui è situato il gene difettoso che causa la neuropatia ottica ereditaria di Leber.