


A cardiomyopathy is a primary disorder of the heart muscle (see also Overview of Cardiomyopathies.)
Hypertrophic cardiomyopathy is a common cause of sudden death in young athletes. It may cause unexplained syncope and may not be diagnosed before autopsy.
Etiology of Hypertrophic Cardiomyopathy
Most cases of hypertrophic cardiomyopathy are inherited. At least 1,500 different mutations that are inherited in an autosomally dominant pattern have been identified; spontaneous mutations can also occur. Prevalence is 1:200 to 1:500 (1); phenotypic expression varies markedly.
Rarely, hypertrophic cardiomyopathy is acquired. It may develop in patients with acromegaly, pheochromocytoma, or neurofibromatosis.
Etiology reference
1. Maron BJ, Desai MY, Nishimura RA, et al. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol 2022;79(4):372-389. doi:10.1016/j.jacc.2021.12.002
Pathophysiology of Hypertrophic Cardiomyopathy
The myocardium is abnormal with cellular and myofibrillar disarray, although this finding is not specific for hypertrophic cardiomyopathy.
In the most common phenotype, the anterior septum and contiguous anterior free wall below the aortic valve are markedly hypertrophied and thickened, with little or no hypertrophy of the left ventricular (LV) posterior wall. Sometimes isolated apical hypertrophy occurs; however, virtually any asymmetric pattern of left ventricular hypertrophy can be observed, and in a small minority of patients even symmetric hypertrophy has been noted.
About two thirds of patients exhibit obstructive physiology at rest or during exercise. Obstruction is the result of mechanical impedance to LV outflow during systole due to systolic anterior motion (SAM) of the mitral valve. During SAM, the mitral valve and valve apparatus are sucked into the LV outflow tract by a Venturi effect of high-velocity blood flow, resulting in obstruction of flow and decrease in cardiac output. Mitral regurgitation can also occur as the result of distortion of leaflet motion by SAM of the mitral valve. This obstruction and valvular regurgitation contribute to the development of symptoms related to heart failure. Less commonly, midventricular hypertrophy leads to an intracavitary gradient at the papillary muscle level, rarely resulting in increased LV wall stress and increased risk of LV apical aneurysm.
Contractility is grossly normal, resulting in a normal ejection fraction (EF). Later, EF is elevated because the ventricle has a small volume and empties nearly completely to maintain cardiac output.
Hypertrophy results in a stiff, noncompliant chamber (usually the left ventricle) that resists diastolic filling, elevating end-diastolic pressure and thus increasing pulmonary venous pressure. As resistance to filling increases, cardiac output decreases, an effect worsened by any outflow tract gradient present. Because tachycardia allows less time for filling, symptoms tend to appear mainly during exercise or tachyarrhythmias. (See also Heart failure with preserved ejection fraction.)
Coronary blood flow may be impaired, causing angina pectoris, syncope, or arrhythmias in the absence of epicardial coronary artery disease (CAD). Flow may be impaired because capillary density relative to myocyte size is inadequate (capillary/myocyte imbalance) or lumen diameter of intramyocardial coronary arteries is narrowed by intimal and medial hyperplasia and hypertrophy. A supply-demand mismatch also may be present due to increased oxygen demand caused by the hypertrophy and adverse loading conditions.
In some cases, myocytes gradually die, probably because capillary/myocyte imbalance causes chronic diffuse ischemia. As myocytes die, they are replaced by diffuse fibrosis. Then, the hypertrophied ventricle with diastolic dysfunction gradually dilates and systolic dysfunction develops.
Symptoms and Signs of Hypertrophic Cardiomyopathy
Typically, symptoms appear between ages 20 and 40 years and are exertional, but symptoms may be highly variable. They include dyspnea, chest pain (usually resembling typical angina), palpitations, and syncope. Because systolic function is preserved, fatigability is seldom reported. The abnormal diastolic function is responsible for most symptoms. In patients with outflow tract obstruction, differentiation of symptoms due to the obstruction versus those caused by abnormal diastolic function can be difficult.
Syncope may occur during exertion either because outflow obstruction worsens with increased contractility or because of ventricular or atrial arrhythmia. Syncope is a marker of increased risk of sudden death.
Blood pressure and heart rate are usually normal, and signs of increased venous pressure are rare. When the outflow tract is obstructed, the carotid pulse has a brisk upstroke, bifid peak, and rapid downstroke. The apex beat may have a sustained thrust due to LV hypertrophy. A 4th heart sound (S4) is often present and is associated with a forceful atrial contraction against a poorly compliant left ventricle in late diastole.
Diagnosis of Hypertrophic Cardiomyopathy
Clinical suspicion (syncope, other symptoms, or murmur)
Echocardiography and/or MRI
Diagnosis is suspected based on a typical murmur and symptoms. Suspicion is increased if the patient has a history of unexplained syncope or a family history of unexplained sudden death. Unexplained syncope in young athletes should always raise suspicion. Hypertrophic cardiomyopathy must be distinguished from aortic stenosis and coronary artery disease, which cause similar symptoms. Less common infiltrative cardiac disorders such as Anderson-Fabry disease and amyloid heart disease may mimic findings of hypertrophic cardiomyopathy.
ECG and 2-dimensional echocardiography and/or MRI (the best noninvasive confirmatory tests) are done. Chest x-ray is often done but is usually normal because the ventricles are not dilated (although the left atrium may be enlarged). Exercise testing and 24-hour ambulatory monitoring may be helpful for patients considered at high risk, although accurately identifying such patients is difficult. Patients with syncope or sustained arrhythmias should be evaluated as inpatients.

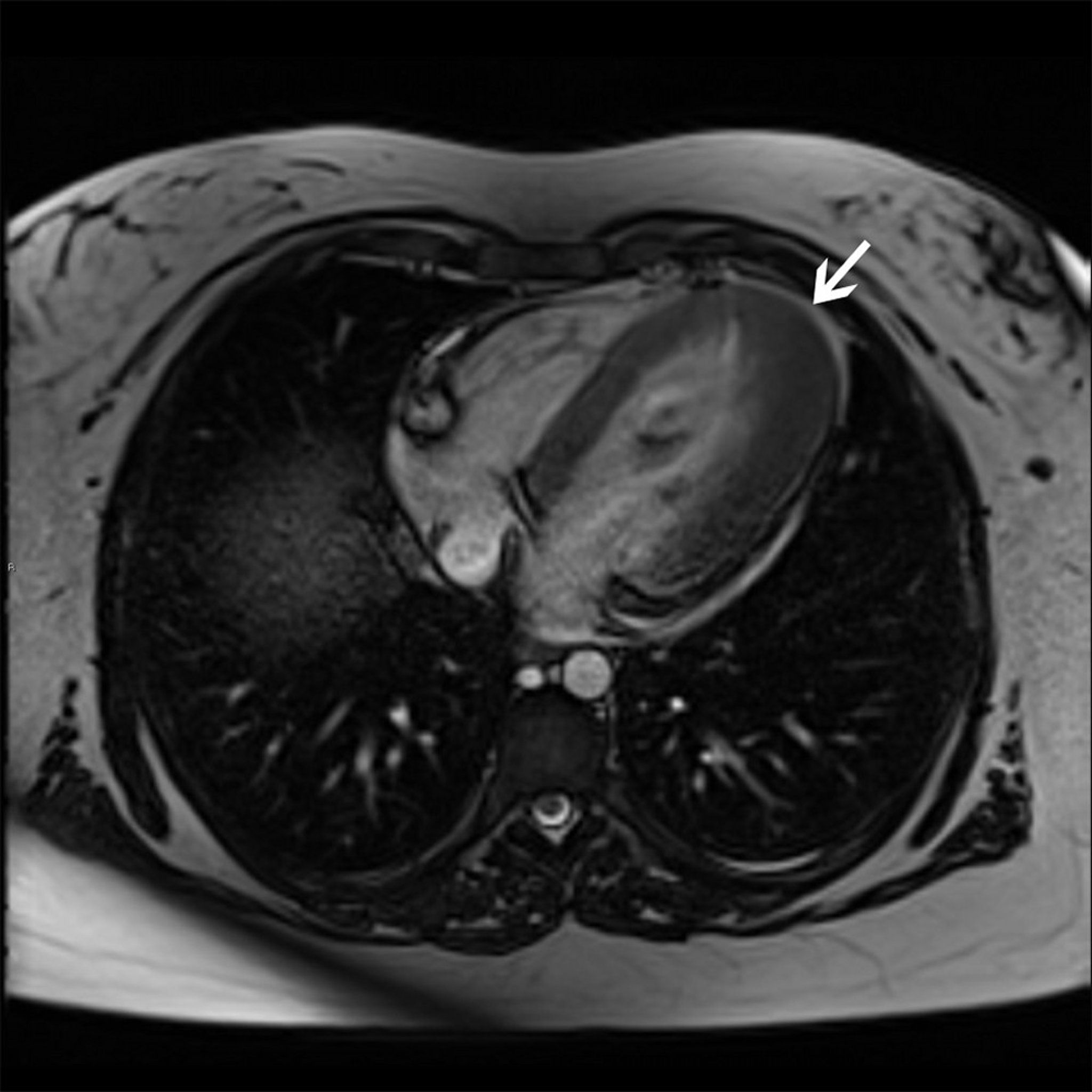
© 2017 Elliot K. Fishman, MD.
The ECG usually shows voltage criteria for LV hypertrophy (eg, S wave in lead V1 plus R wave in lead V5 or V6 > 35 mm). Very deep septal Q waves in leads I, aVL, V5, and V6 are often present with asymmetric septal hypertrophy; hypertrophic cardiomyopathy sometimes produces a QRS complex in V1 and V2, simulating previous septal infarction. T waves are usually abnormal; the most common finding is deep symmetric T-wave inversion in leads I, aVL, V5, and V6. ST-segment depression in the same leads is common (particularly in the apical obliterative form). The P wave is often broad and notched in leads II, III, and aVF, with a biphasic P wave in leads V1 and V2, indicating left atrial hypertrophy. Incidence of preexcitation phenomenon of the Wolff-Parkinson-White syndrome type, which may cause palpitations, is increased. Bundle branch block is common.
Two-dimensional Doppler echocardiography can differentiate the forms of cardiomyopathy (see figure Forms of Cardiomyopathy) and quantify the severity of hypertrophy and degree of outflow tract obstruction. These measurements are particularly useful for monitoring the effect of medical or surgical treatment. Midsystolic closure of the aortic valve sometimes occurs when outflow tract obstruction is severe. Ambulatory 24-hour monitoring is recommended during the initial evaluation and every 1 to 3 years to assess risk of sudden cardiac death and help guide treatment of arrhythmias (1).
Cardiac catheterization is usually done only when invasive therapy is considered. Usually, no significant stenoses are present in the coronary arteries, but older patients may have coexisting coronary artery disease.

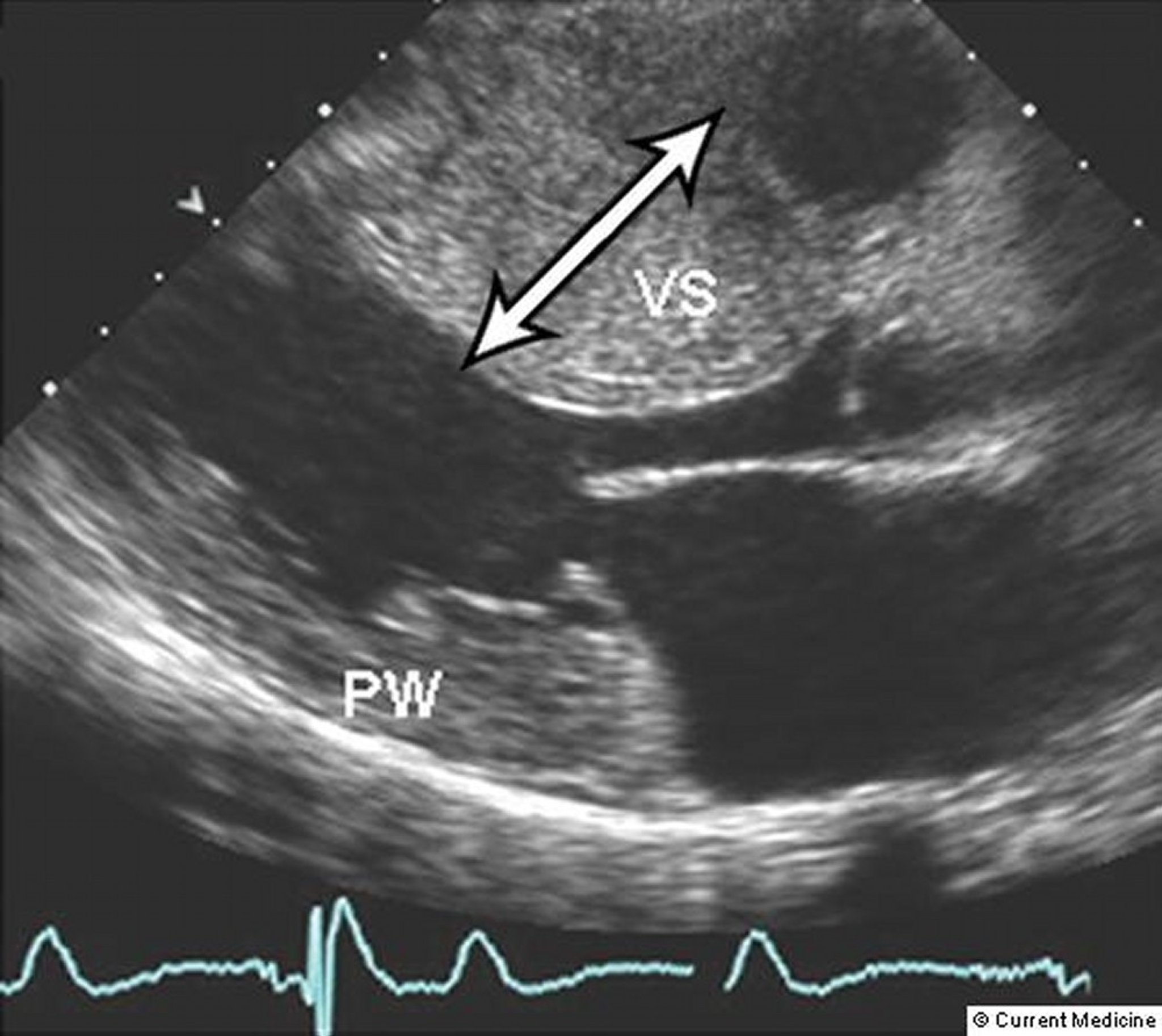
© Springer Science+Business Media
Genetic markers do not identify high-risk individuals or influence treatment. However, genetic testing may be of benefit in screening family members.
Diagnosis reference
1. Maron BJ, Desai MY, Nishimura RA, et al. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol 2022;79(4):372-389. doi:10.1016/j.jacc.2021.12.002
Treatment of Hypertrophic Cardiomyopathy
Beta-blockers
Rate-limiting and negative inotropic calcium channel blockers
Avoidance of nitrates, diuretics, and angiotensin-converting enzyme (ACE) inhibitors
Possibly implantable cardioverter-defibrillator and sometimes surgery or ablative procedures
Disopyramide appears to be most effective for patients with a resting gradient whereas beta-blockers are best at blunting the gradient that occurs during exercise. Beta-blockers are generally considered first-line treatment for patients with the obstructive phenotype; disopyramide helps relieve symptoms for patients in whom first-line therapy with beta-blockers, verapamil, or diltiazem has failed.
Patients who continue to experience symptoms related to significant outflow tract gradients (≥ 50 mm Hg) despite medical therapy are candidates for invasive treatment. When done at an experienced center, surgical myectomy has a low operative mortality with excellent outcomes, making it the preferred therapy in such patients. Percutaneous catheter alcohol septal ablation is an alternative to surgery in older patients and others who are at high surgical risk.
Medications that reduce preload (eg, nitrates, diuretics, ACE inhibitors, angiotensin II1, 2).
If syncope or sudden cardiac arrest has occurred or if sustained ventricular arrhythmia is confirmed by ECG or 24-hour ambulatory monitoring, an implantable cardioverter-defibrillator (ICD) should usually be placed. Controversy exists regarding the need to place a defibrillator in patients without syncope, sudden cardiac arrest, or ventricular arrhythmias. It is generally believed that ICD insertion should be considered in patients with high-risk features (3). High-risk features include
A family history of premature sudden cardiac arrest
Left ventricular wall thickness > 3 cm
Delayed enhancement on cardiac MRI
Unexplained syncope
Survival after a cardiac arrest due to ventricular arrhythmia
Multiple burst of nonsustained ventricular tachycardia (NSVT)
Left ventricular ejection fraction (LVEF) < 50% (end-stage disease)
Left ventricular aneurysm
Myomectomy and alcohol ablation have been used to decrease outflow obstruction and relive symptoms. There is no proven difference on mortality between these interventions, but myomectomy is more effective for relieving obstruction and decreasing the need for repeat interventions (4).
Avoidance of competitive sports was previously recommended because sudden deaths can occur during increased exertion. Current guidelines recommend that athletes with HCM undergo comprehensive evaluation and shared discussion of potential risk with an expert HCM specialist.
Treatment of the dilated congestive phase of hypertrophic cardiomyopathy is the same as that of dilated cardiomyopathy with predominant systolic dysfunction.
Genetic counseling is appropriate for patients with asymmetric septal hypertrophy.
Treatment references
1. Olivotto I, Oreziak A, Barriales-Villa R, et al: Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 396:759–769, 2020. doi: 10.1016/S0140-6736(20)31792-X
2. Spertus JA, Fine JT, Elliott P, et al: Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): health status analysis of a randomized, double-blind, placebo-controlled, phase 3 trial. Lancet 397:2467–2475, 2012. doi: 10.1016/S0140-6736(21)00763-7
3. Maron MS, Rowin EJ, Wessler BS, et al: Enhanced American College of Cardiology/American Heart Association Strategy for Prevention of Sudden Cardiac Death in High-Risk Patients With Hypertrophic Cardiomyopathy. JAMA Cardiol 4(7):644–657, 2019. doi:10.1001/jamacardio.2019.1391
4. Nguyen A, Schaff HV, Hang D, et al: Surgical myectomy versus alcohol septal ablation for obstructive hypertrophic cardiomyopathy: A propensity score-matched cohort. J Thorac Cardiovasc Surg 157(1):306–315.e3, 2019. doi:10.1016/j.jtcvs.2018.08.062
Prognosis for Hypertrophic Cardiomyopathy
Overall, annual mortality is about 1% for adults but is higher for children. Death is usually sudden, and sudden death is the most common sequela; chronic heart failure occurs less often. A higher risk of sudden cardiac death is predicted by the presence of the following risk factors:
Family history of sudden cardiac death due to hypertrophic cardiomyopathy, cardiac arrest, or sustained ventricular arrhythmias
Personal history of unexplained syncope, cardiac arrest, or sustained ventricular arrhythmias
Multiple repetitive non-sustained ventricular tachycardia (on ambulatory ECG)
Massive left-ventricular hypertrophy (thickness ≥ 30 mm), LV dysfunction (EF < 50%) , LV apical aneurysm
Extensive and diffuse late gadolinium enhancement on MRI
Key Points
Hypertrophic cardiomyopathy is usually due to one of numerous genetic mutations that cause various types of ventricular hypertrophy that restrict filling (ie, cause diastolic dysfunction) and sometimes obstruct LV outflow.
Coronary blood flow may be impaired even in the absence of coronary artery atherosclerosis because capillary density is inadequate and the intramyocardial coronary arteries are narrowed by intimal and medial hyperplasia and hypertrophy.
At a young age, patients may have chest pain, dyspnea, palpitations, syncope, and sometimes sudden death, typically triggered by exertion.
Echocardiography is done, but, if available, MRI best shows the abnormal myocardium.
Avoid nitrates and other medications that decrease preload (eg, diuretics, angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers) because these decrease left ventricular size and worsen left ventricular function.
Place an implantable cardioverter-defibrillator for patients with syncope or sudden cardiac arrest.
Do surgical myectomy or alcohol septal ablation in patients with symptoms despite medical therapy.
More Information
The following English-language resource may be useful. Please note that THE MANUAL is not responsible for the content of this resource.
American Heart Association Guidelines on Hypertrophic Cardiomyopathy: Ommen SR, Mital S, Burke MA, et al: 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines [published correction appears in Circulation. 2020 Dec 22;142(25):e633]. Circulation 20 November 2020. doi: 10.1161/CIR.0000000000000937






