


Ventilatory failure is a rise in PaCO2 (hypercapnia) that occurs when the respiratory load can no longer be supported by the strength or activity of the system. The most common causes are severe acute exacerbations of asthma and chronic obstructive pulmonary disease (COPD), overdoses of drugs that suppress ventilatory drive, and conditions that cause respiratory muscle weakness (eg, Guillain-Barré syndrome, myasthenia gravis, botulism). Findings include dyspnea, tachypnea, and confusion. Death can result. Diagnosis is by arterial blood gas measurement and patient observation; chest x-ray and clinical evaluation may help delineate cause. Treatment varies by condition but often includes mechanical ventilation.
(See also Overview of Mechanical Ventilation.)
The 2 most common causes of ventilatory failure are
Severe acute exacerbation of asthma (ie, status asthmaticus)
Exacerbation of COPD (chronic obstructive pulmonary disease)
Respiratory failure due to COPD is termed acute-on-chronic respiratory failure (ACRF).
Pathophysiology of Ventilatory Failure
Hypercapnia occurs when alveolar ventilation either falls or fails to rise adequately in response to increased carbon dioxide production. A fall in alveolar ventilation results from a decrease in minute ventilation or an increase in dead space ventilation without appropriate compensation by increasing minute ventilation.
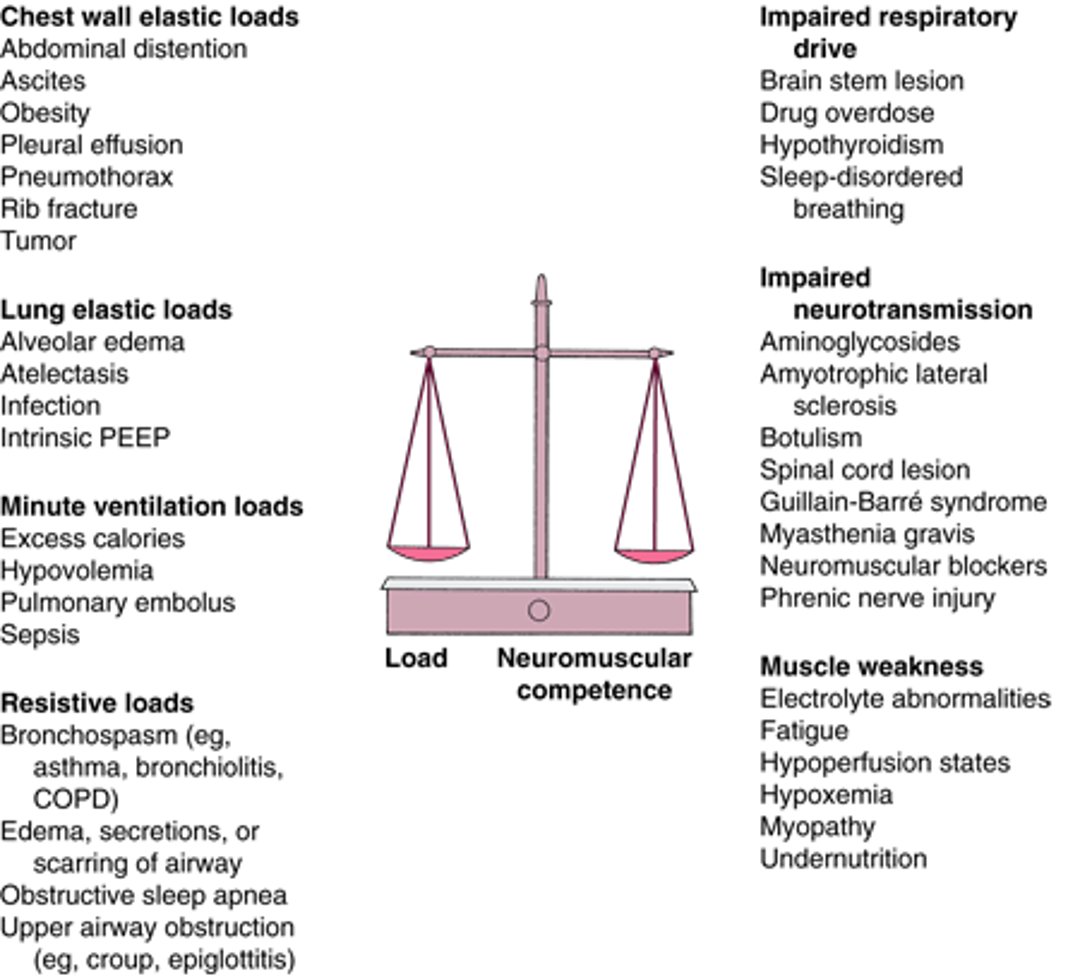
Ventilatory failure can occur when there is excessive load on the respiratory system (eg, resistive loads or lung and chest wall elastic loads) versus neuromuscular competence for an effective inspiratory effort. When the minute ventilation load increases (eg, as occurs in sepsis), a compromised respiratory system may not be able to meet this increased demand (for causes, see figure The balance between load and neuromuscular competence).
Physiologic dead space is the part of the respiratory tree that does not participate in gas exchange. It includes
Anatomic dead space (oropharynx, trachea, and airways)
Alveolar dead space (ie, alveoli that are ventilated but not perfused)
Physiologic dead space can also result from shunt or low ventilation/perfusion (V/Q) if patients cannot increase their minute ventilation appropriately. The physiologic dead space normally is about 30 to 40% of tidal volume but increases to 50% in intubated patients and to >70% in massive pulmonary embolism, severe emphysema, and status asthmaticus. Thus, for any given minute ventilation, the greater the dead space, the poorer the carbon dioxide elimination.
Increased carbon dioxide production, as occurs with fever, sepsis, trauma, burns, hyperthyroidism, and malignant hyperthermia, is not a primary cause of ventilatory failure because patients should increase their ventilation to compensate. Ventilatory failure associated with these problems results only when the ability to compensate is compromised.
Hypercapnia lowers arterial pH (respiratory acidosis). Severe acidemia (pH < 7.2) contributes to pulmonary arteriolar vasoconstriction, systemic vascular dilation, reduced myocardial contractility, hyperkalemia, hypotension, and cardiac irritability, with the potential for life-threatening arrhythmias. Acute hypercapnia also causes cerebral vasodilation and increased intracranial pressure, a major problem in patients with acute head injury. Over time, tissue buffering and renal compensation can largely correct the acidemia. However, sudden increases in PaCO2 (partial pressure of carbon dioxide) can occur faster than compensatory changes (PaCO2 rises 3 to 6 mm Hg/minute in a totally apneic patient).
The balance between load (resistive, elastic, and minute ventilation) and neuromuscular competence (drive, transmission, and muscle strength) determines the ability to sustain alveolar ventilation
PEEP = positive end-expiratory pressure. |

Symptoms and Signs of Ventilatory Failure
The predominant symptom of ventilatory failure is dyspnea.
When ventilatory failure is due to increased load, signs include vigorous use of accessory ventilatory muscles, tachypnea, tachycardia, diaphoresis, anxiety, declining tidal volume, irregular or gasping breathing patterns, and paradoxical abdominal motion. When it is due to impaired respiratory drive, signs include hypopnea and/or low respiratory rates will be evident.
Ventilatory failure results in hypercapnia, causing central nervous system manifestations ranging from subtle personality changes to marked confusion, obtundation, or coma. Chronic hypercapnia is better tolerated than acute and has fewer symptoms.
Diagnosis of Ventilatory Failure
Arterial blood gas (ABGs) measurement
Chest x-ray
Tests to determine etiology
Ventilatory failure should be suspected in patients with respiratory distress, visible ventilatory fatigue or cyanosis, or changes in sensorium and in those with disorders causing neuromuscular weakness. Tachypnea is also a concern; respiratory rates > 28 to 30/minute cannot be sustained for very long, particularly in older or weakened patients.
If ventilatory failure is suspected, ABG analysis, continuous pulse oximetry, and a chest x-ray should be done. Respiratory acidosis revealed by the ABG measurement (eg, pH < 7.35 and PCO2 > 50) confirms the diagnosis. Patients with chronic ventilatory failure often have quite elevated PCO2 (eg, 60 to 90 mm Hg) at baseline, typically with a pH that is only slightly acidemic. In such patients, the degree of acidemia rather than the PCO2 must serve as the primary marker for acute hypoventilation.
Because ABG measurements can be normal or show insufficient respiratory compensation in patients with metabolic acidosis and incipient ventilatory failure, certain bedside pulmonary function tests can help predict ventilatory failure, particularly in patients with neuromuscular weakness who may succumb to ventilatory failure without exhibiting respiratory distress. Vital capacity < 10 to 15 mL/kg and an inability to generate a negative inspiratory force of 15 cm H2O suggest imminent ventilatory failure.
Once ventilatory failure is diagnosed, the cause must be identified. Sometimes a known ongoing disorder (eg, coma, acute asthma exacerbation, COPD exacerbation, severe hypothyroidism, myasthenia gravis, botulism) is an obvious cause. In other cases, history is suggestive; sudden onset of tachypnea and hypotension after surgery suggests pulmonary embolism, and focal neurologic findings suggest a central nervous system or neuromuscular cause. Neuromuscular competence may be assessed through measurement of inspiratory muscle strength (negative inspiratory force and positive expiratory force), neuromuscular transmission (nerve conduction tests and electromyography), and investigations into causes of diminished drive (toxicology screens, brain imaging, and thyroid function tests).
Treatment of Ventilatory Failure
Treatment of cause
Often positive pressure ventilation
Treatment of ventilatory failure aims to correct the imbalance between the strength of the respiratory system and its load and varies with etiology. Obvious precipitants (eg, bronchospasm, mucus plugging, foreign bodies) should be corrected if possible.
Status asthmaticus
Patients with status asthmaticus should be treated in an intensive care unit by personnel skilled in airway management. (See also Treatment of Acute Asthma Exacerbations.)
Noninvasive positive pressure ventilation (NIPPV) can immediately reduce the work of breathing and may forestall endotracheal intubation until drug therapy can take effect. The mask introduction must be done carefully to enhance comfort by perhaps starting with titration of expiratory positive airway pressure (EPAP) alone because one of the major functions of inspiratory positive airway pressure (IPAP) is to increase tidal volume, and, in these patients, end-expiratory lung volume approaches total lung capacity (see also Respiratory Mechanics). After an explanation of its benefit, patients hold the mask against their face while modest amounts of pressure are applied (continuous positive airway pressure [CPAP] 3 to 5 cm H2O). Once tolerated, the mask is strapped in place while pressures are increased to patient comfort and reduced work of breathing as assessed by respiratory rate and accessory muscle use. Patients should be selected carefully, and settings should be adjusted on an individual basis.
Conventional mechanical ventilation via endotracheal intubation is indicated for impending respiratory failure as indicated clinically by signs such as obtundation, monosyllabic speech, slumped posture, and shallow breathing. ABG values that reveal worsening hypercapnia are also an indication, although blood gas confirmation is not required and should not replace the physician’s judgment. Oral intubation is preferred over nasal because a larger endotracheal tube, which decreases airway resistance and permits easier suctioning, can be used.
Hypotension and pneumothorax occasionally occur after intubation for status asthmaticus and acute exacerbations of COPD (see also Complications of Mechanical Ventilation and Safeguards). These complications and their corresponding mortality have declined significantly because of a ventilator strategy that emphasizes limiting dynamic hyperinflation over achieving eucapnia. In status asthmaticus, ventilation sufficient to achieve a normal pH typically causes severe hyperinflation. To avoid hyperinflation, initial ventilator settings include a tidal volume of 5 to 7 mL/kg and a respiratory rate of 10 to 18/minute. Inspiratory flows may need to be quite high (eg, 70 to 120 L/minute) with a square wave pattern to facilitate maximum time in exhalation. Dangerous dynamic hyperinflation is unlikely so long as the measured plateau pressure is < 30 to 35 cm H2O and intrinsic positive end-expiratory pressure (intrinsic PEEP) is < 15 cm H2O (although these pressures may be difficult to measure because of inspiratory and expiratory respiratory muscle activity). Plateau pressure > 35 cm H2O is managed by reducing the tidal volume (assuming that clinical evaluation does not indicate that the high pressures are the result of decreased compliance of the chest wall or abdomen) or the respiratory rate.
Although it is possible to reduce peak airway pressure by reducing peak flow rate or by changing the flow waveform to a descending profile (ie, so that the flow rate is high at the beginning of the breath and decreased over time), such changes should not be made. Although high flow rates require a high pressure to overcome the high airway resistance of status asthmaticus, this pressure is dissipated across robust, cartilage-containing airways. Lower flow rates (eg, < 60 L/minute) reduce time available for exhalation, thereby increasing the end-expiratory volume (and the resultant intrinsic PEEP) and allowing a greater inspiratory volume during the next breath. Occasionally, patients with high intrinsic PEEP may need to have PEEP increased on the ventilator to facilitate triggering and reduce inspiratory work of breathing.
Using low tidal volumes often results in hypercapnia, which is permitted for the greater good of reducing dynamic hyperinflation. An arterial pH > 7.15 is generally well tolerated physiologically but often requires large doses of sedatives and opioids. Neuromuscular blockers should be avoided after the peri-intubation period because use of these agents in combination with corticosteroids can cause a severe and occasionally irreversible myopathy, particularly after 24 hours of combined use. Patient agitation should be managed with sedation rather than paralysis, but ideally ventilation can be adjusted to patients’ needs so as to reduce the need for sedation.
Most patients with status asthmaticus improve to the point of liberation from mechanical ventilation within 2 to 5 days, although a minority experience protracted severe airflow obstruction. See Liberation from Mechanical Ventilation for a discussion on the general approach.
Acute-on-chronic respiratory failure
In patients with acute-on-chronic respiratory failure (ACRF) caused by COPD, the oxygen cost of breathing is several times that of patients without underlying lung disease. This increased respiratory load occurs in the setting of barely adequate neuromuscular competence, so patients easily become too tired to maintain ventilation. These patients are vulnerable to respiratory failure from seemingly trivial insults, and recovery requires systematic identification and correction of these precipitants (see also Treatment of Acute COPD Exacerbation). To restore the balance between neuromuscular competence and load, clinicians reduce airflow obstruction and dynamic hyperinflation with bronchodilators and corticosteroids and treat infection with antibiotics. Low serum levels of potassium, phosphorus, and magnesium may exacerbate muscle weakness, frustrating recovery, and must be identified and treated.
NIPPV is the preferred initial treatment for many patients with ACRF, resulting in decreased rates of ventilator-associated pneumonia, length of stay, and mortality compared with endotracheal intubation. Perhaps 75% of patients managed with NIPPV do not require endotracheal intubation. Advantages include the ease of application and removal; once initial stabilization has occurred, NIPPV may be stopped temporarily to allow oral intake in selected patients. Trials of unassisted breathing are easily done, and NIPPV can be reapplied as indicated.
Settings should be adjusted to the work of breathing as assessed by patient report, respiratory rate and tidal volume, and accessory muscle use. In many patients, EPAP alone may be sufficient, which is beneficial because one of the major functions of inspiratory positive airway pressure is to increase tidal volume, and, in these patients, end-expiratory lung volume approaches total lung capacity (see also Respiratory Mechanics). Deterioration (and need for endotracheal intubation) is best assessed clinically; ABG measurements may be misleading. Although worsening hypercapnia typically indicates treatment failure, patients differ markedly in tolerance of hypercapnia. Some patients with PaCO2> 100 mm Hg are alert and conversant on NIPPV, whereas others require intubation at much lower levels.
Conventional mechanical ventilation in ACRF aims to minimize dynamic hyperinflation and counter the adverse effects of intrinsic PEEP while resting the fatigued respiratory muscles. Initial recommended settings are assist-control (A/C) with a tidal volume of 5 to 7 mL/kg and a respiratory rate of 20 to 24/minute, although some patients need lower initial rates to limit intrinsic PEEP. This intrinsic PEEP represents an inspiratory threshold load that must be overcome by the patient to trigger the ventilator, further increasing the work of breathing and preventing full rest on the ventilator. To counterbalance the effect of intrinsic PEEP, external PEEP should be applied to a level ≤ 85% of intrinsic PEEP (typical setting 5 to 10 cm H2O). This application decreases the inspiratory work of breathing without increasing dynamic hyperinflation. High inspiratory flow rates should be used to maximize the time for expiration. These settings minimize the risk of alkalemia that follows overly vigorous initial ventilation. Hypotension may also occur immediately after intubation (see also Complications of Mechanical Ventilation and Safeguards).
Most patients require full ventilatory support for 24 to 48 hours before spontaneous breathing trials are considered. It has not been determined whether this duration of treatment is needed to rest the respiratory muscles or to allow hyperinflation to diminish, thereby increasing respiratory muscle strength. The patient often sleeps heavily during this time and, in contrast to patients with asthma, typically requires little sedation. Adequate rest is often not achieved unless sufficient attention is paid to ongoing patient effort. This effort may manifest as accessory muscle use, inappropriately low airway pressures at the onset or throughout inspiration, or frequent failures to trigger the ventilator, indicating high intrinsic PEEP, weakness, or both.






