


Abnormal gene coding for retinal proteins appears to be the cause of retinitis pigmentosa; several genes have been identified. Transmission may be autosomal recessive, autosomal dominant, or, infrequently, X-linked. It may occur as part of a syndrome (eg, Bassen-Kornzweig, Laurence-Moon). One of these syndromes includes congenital hearing loss as well (Usher syndrome).
Symptoms and Signs of Retinitis Pigmentosa
Retinal rods are affected, causing defective night vision that becomes symptomatic at varying ages, sometimes in early childhood. Night vision may eventually be lost. A peripheral ring scotoma (detectable by visual field testing) widens gradually, and central vision may also be affected in advanced cases. Vision decreases as the macula becomes increasingly involved and can evolve to legal blindness.
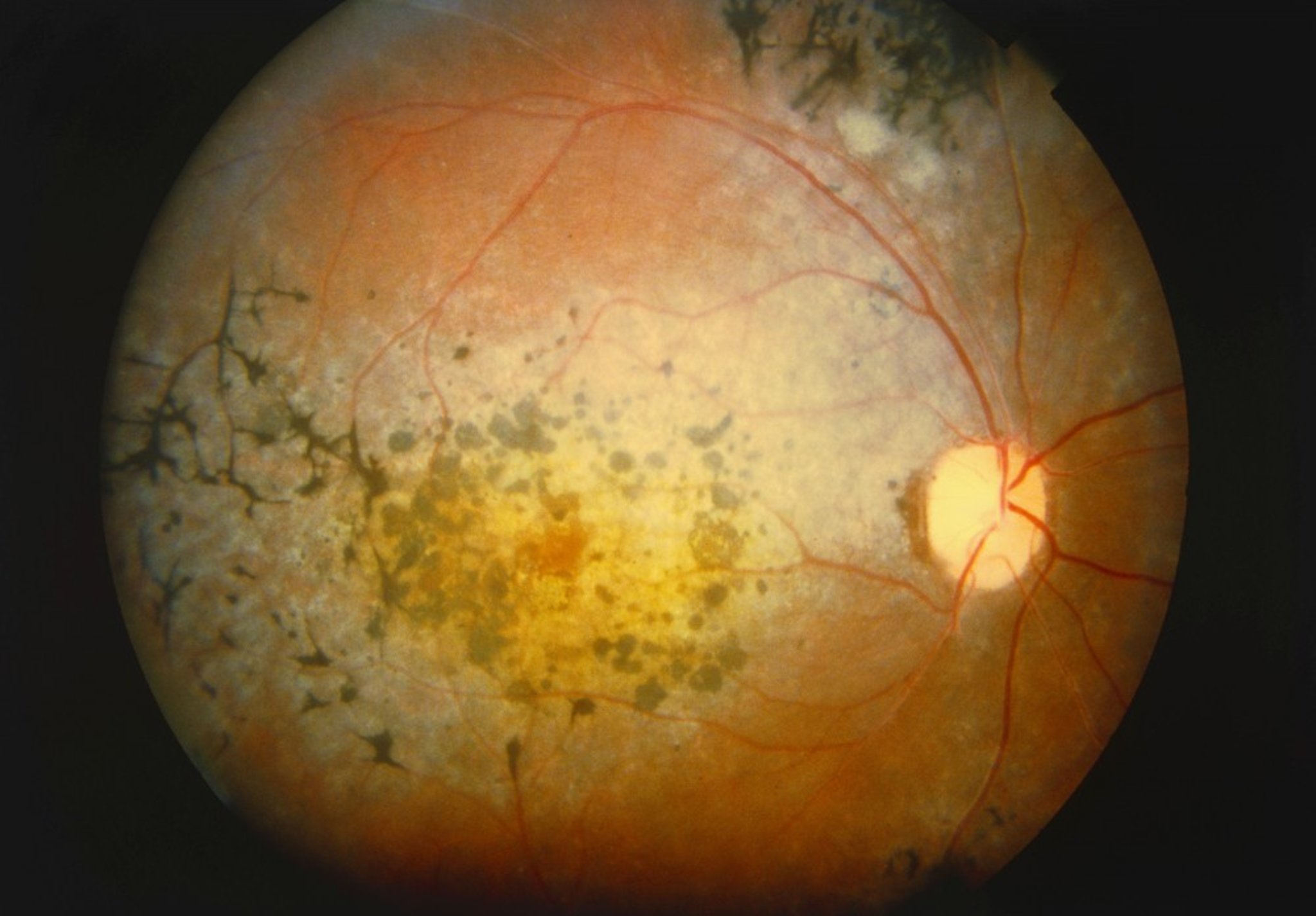
Hyperpigmentation in a bone-spicule configuration in the midperipheral retina is the most conspicuous funduscopic finding. Other findings include the following:
Narrowing of the retinal arterioles
Cystoid macular edema
Waxy yellow appearance of the disk
Posterior subcapsular cataracts
Cells in the vitreous (less common)
Myopia
Diagnosis of Retinitis Pigmentosa
Funduscopy
Electroretinography

WESTERN OPHTHALMIC HOSPITAL/SCIENCE PHOTO LIBRARY
Family members should be examined and tested as necessary or desired to establish the hereditary pattern. Patients with a hereditary syndrome may wish to seek genetic counseling before having children.
Treatment of Retinitis Pigmentosa
Omega-3 fatty acids
Lutein plus zeaxanthin
Carbonic anhydrase inhibitors for cystoid macular edema
Intraocular computer chip implants
1). This treatment can restore ambulatory vision in these patients. For patients with total or near total vision loss, epiretinal and subretinal computer chip implants can restore some visual sensations.
Treatment reference
1. Maguire AM, Russell S, Wellman JA, et alOphthalmology 126(9):1273-1285, 2019. doi: 10.1016/j.ophtha.2019.06.017
Key Points
Early symptoms of retinitis pigmentosa include defective night vision and peripheral vision.
Diagnose by hyperpigmentation in a bone-spicule configuration on funduscopy and confirm with electroretinography.
Treat patients with cystoid macular edema with carbonic anhydrase inhibitors.






