


A number of connective tissue diseases cause inflammation of the uveal tract. (See also Overview of Uveitis.)
Spondyloarthropathies
The seronegative spondyloarthropathies are a common cause of anterior uveitis.
Among the seronegative spondyloarthropathies, ocular inflammation is most common with ankylosing spondylitis but also occurs with reactive arthritis, inflammatory bowel disease (ulcerative colitis and Crohn disease), and psoriatic arthritis. Uveitis is classically unilateral, but recurrences are common and active inflammation may alternate between eyes. Men are affected more commonly than women. Most patients, regardless of sex, are HLA-B27 positive.
Juvenile idiopathic arthritis (JIA)
JIA, previously known as juvenile RA, characteristically causes chronic bilateral iridocyclitis in children, particularly those with the pauciarticular variety. Unlike most forms of anterior uveitis, however, JIA-associated uveitis tends not to cause pain, photophobia, and conjunctival injection but only blurring and miosis and is, therefore, often referred to as white iritis. It can be asymptomatic. JIA-associated uveitis is more common among girls. Because symptoms can be overlooked or absent, patients with JIA should be regularly screened.
Rheumatoid arthritis, in contrast, is not associated with isolated uveitis but can cause scleritis, which may cause secondary uveal tract inflammation.
Sarcoidosis
Sarcoidosis accounts for 10 to 20% of cases of uveitis, and about 25% of patients with sarcoidosis develop uveitis. Sarcoid uveitis is more common among patients of African descent and older patients.
Virtually any symptoms and signs of anterior, intermediate, posterior, or panuveitis can occur. Suggestive findings include conjunctival granulomas, large keratic precipitates on the corneal endothelium (so-called granulomatous or mutton fat precipitates), iris granulomas, and retinal vasculitis. Biopsy of suggestive lesions, which provides the most secure diagnosis, is usually done on the conjunctiva; it is rarely done on intraocular tissues because of the risk associated with the procedure.

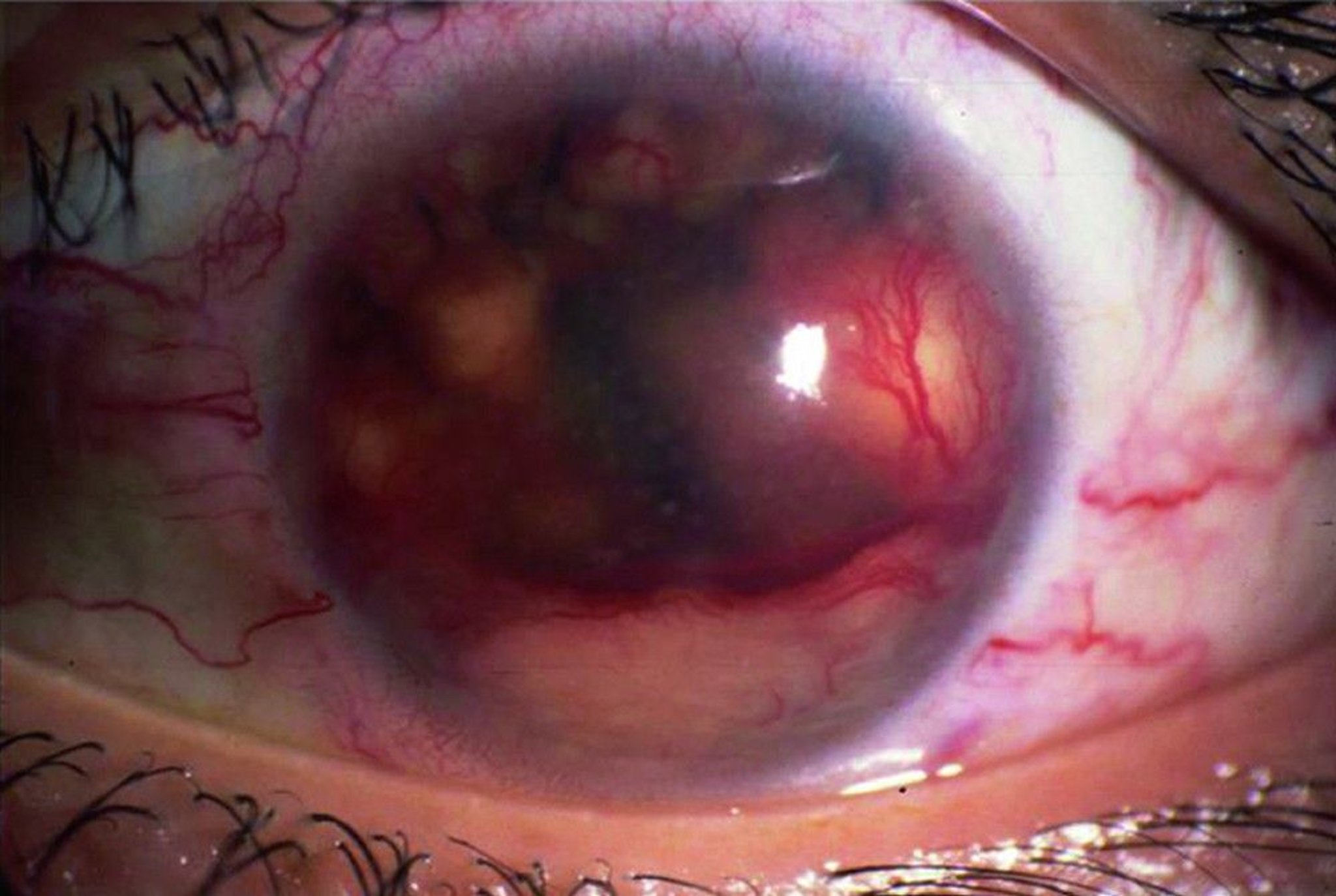
© Springer Science+Business Media
Behçet disease
Behçet disease is rare in North America but is a fairly common cause of uveitis in the Middle East and Far East.
Typical findings include severe anterior uveitis with hypopyon, retinitis, retinal vasculitis, and optic disk inflammation. The clinical course is usually severe with multiple recurrences.
Diagnosis requires the presence of associated systemic manifestations, such as oral aphthous or genital ulcers; dermatitis, including erythema nodosum; thrombophlebitis; or epididymitis. Oral aphthae may be biopsied to show an occlusive vasculitis. There are no laboratory tests for Behçet disease, but it is associated with HLA-B51.

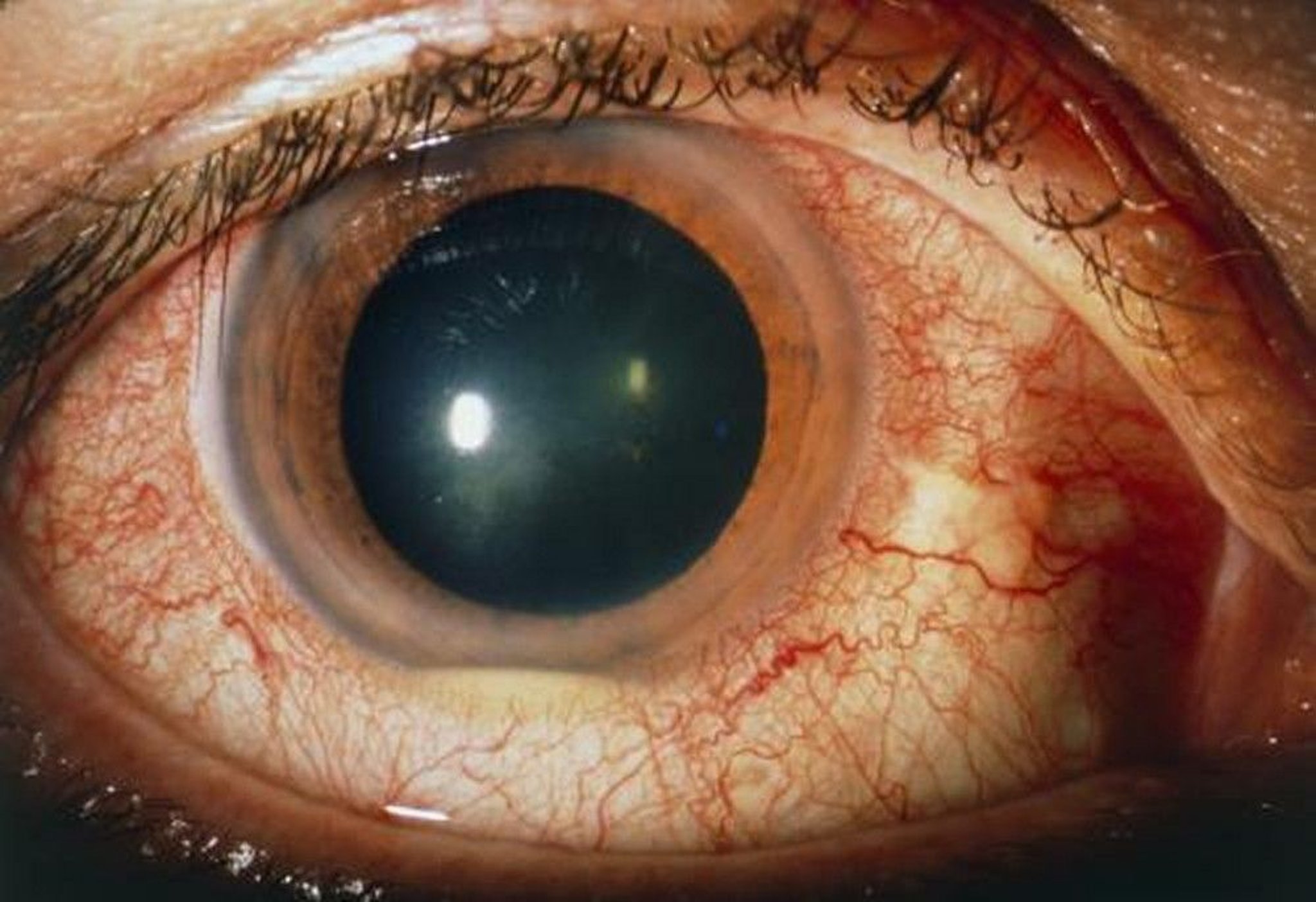
SUE FORD/SCIENCE PHOTO LIBRARY
Vogt-Koyanagi-Harada (VKH) disease
Vogt-Koyanagi-Harada disease is an uncommon systemic disorder characterized by uveitis accompanied by cutaneous and neurologic abnormalities. VKH disease is particularly common among people of Asian, Asian Indian, American Indian, and Hispanic descent. Women in their 20s and 30s are affected more often than men. The etiology is unknown, although an autoimmune reaction directed against melanin-containing cells in the uveal tract, skin, inner ear, and meninges is strongly suspected.
Neurologic symptoms tend to occur early and include tinnitus, dysacusis (auditory agnosia), vertigo, headache, and meningismus. Cutaneous findings frequently occur later and include patchy vitiligo (especially common on the eyelids, low back, and buttocks), poliosis (a localized patch of white hair, which may involve the eyelashes), and alopecia, often involving the head and neck. Common findings include serous retinal detachment, optic disk edema, and choroiditis. Long-term complications include cataracts, glaucoma, subretinal fibrosis, and choroidal neovascularization.
Tubulointerstitial nephritis and uveitis syndrome (TINU)
TINU typically presents with nongranulomatous acute bilateral anterior uveitis, though it is frequently accompanied by posterior complications, including edema of the optic nerve and macula. It most commonly affects adolescent females but can present at any age. Symptoms include eye pain and redness, decreased visual acuity, and photophobia. Patients may have a history of viral prodrome symptoms and also flank pain, polyuria, and nocturia. Evaluation for nephritis may include urine beta-2 microglobulin levels and sometimes renal biopsy. While the acute kidney injury is frequently self-limited in TINU, the ocular disease is often chronic (1).
TINU reference
1. Koreishi AF, Zhou M, Goldstein DA: Tubulointerstitial nephritis and uveitis syndrome: Characterization of clinical features. Ocul Immunol Inflamm 17;29(7-8):1312-1317, 2021. doi: 10.1080/09273948.2020.1736311






