


Sickle cell disease (a hemoglobinopathy
Hemoglobinopathies are genetic disorders affecting the hemoglobin molecule. Hemoglobin S was the first abnormal hemoglobin to be identified. Homozygotes (about 0.3% of people with African ancestry in the United States) have sickle cell anemia; heterozygotes (8 to 13% of people with African ancestry in the United States) are typically not anemic but have a risk of other complications.
Pathophysiology of Sickle Cell Disease
Hemoglobin (Hb) molecules consist of polypeptide chains whose chemical structure is genetically controlled. The normal adult hemoglobin molecule (Hb A) consists of 2 pairs of chains designated alpha and beta. Normal adult blood also contains ≤ 2.5% hemoglobin A2 (composed of alpha and delta chains) and < 1.4% hemoglobin F (fetal hemoglobin), which has gamma chains in the place of beta chains (see also Hemoglobinopathies in Pregnancy). Hemoglobin F predominates during gestation and gradually decreases, particularly in the first months of life; its concentration increases in certain disorders of hemoglobin synthesis and in aplastic anemia, other bone marrow failure disorders, and myeloproliferative neoplasms.
Some hemoglobinopathies result in anemias that are severe in patients who are homozygous but mild in those who are heterozygous. Some patients are compound heterozygotes for 2 different hemoglobinopathies and have anemia of varying severity.
Different hemoglobins (Hbs), as distinguished by electrophoretic mobility, are alphabetically designated in order of discovery (eg, A, B, C), although the first abnormal hemoglobin, sickle cell hemoglobin, was designated Hb S. Structurally different hemoglobins with the same electrophoretic mobility are named for the city or location in which they were discovered (eg, Hb S Memphis, Hb C Harlem). Standard description of a patient’s hemoglobin composition places the hemoglobin of greatest concentration first (eg, AS in sickle cell trait).
In the US, common anemias include those caused by genetic mutations resulting in Hb S or Hb C disease, and the thalassemias. Immigration to the US of people with Southeast Asian ancestry has made Hb E disease common.
In hemoglobin S, valine is substituted for glutamic acid in the 6th amino acid of the beta chain. Oxygenated Hb S is much less soluble than oxygenated Hb A; it forms a semisolid gel that causes red blood cells (RBCs) to deform into a sickle shape at sites of low PO2. Distorted, inflexible RBCs adhere to vascular endothelium and plug small arterioles and capillaries, which leads to infarction. Vaso-occlusion also causes endothelial injury, which results in inflammation and can lead to thromboses. Because sickled RBCs are fragile, the mechanical trauma of circulation causes hemolysis (see Overview of Hemolytic Anemia). Chronic compensatory marrow hyperactivity deforms the bones.
Acute exacerbations
Acute exacerbations (crises) occur intermittently, often for no known reason. In some cases, crisis appears to be precipitated by
Fever
Viral infection
Local trauma
Vaso-occlusive crisis (pain crisis) is the most common type; it is caused by tissue hypoxia and leads to ischemia and infarction, typically in the bones, but also in the spleen, lungs, or kidneys.
Aplastic crisis occurs when bone marrow erythropoiesis slows during acute infection due to human parvovirus, during which an acute erythroblastopenia may occur.
Acute chest syndrome results from pulmonary microvascular occlusion and is a common cause of death, with mortality rates of up to 10%. It occurs in all age groups but is most common in childhood. Repeated episodes predispose to chronic pulmonary hypertension.
Sequestration crisis typically occurs in children whose spleen has not yet become fibrotic due to repeated splenic infarction. Acute sequestration of sickled cells in the spleen exacerbates anemia.
Hepatic sequestration may occur in children or adults, causing right upper quadrant pain. Rapid enlargement of the liver can occur and may be accompanied by intrahepatic cholestasis and renal failure.
Priapism, a serious complication that can cause erectile dysfunction, is most common in young men.
Complications
Chronic spleen damage can lead to autoinfarction and increases susceptibility to infection, particularly pneumococcal and Salmonella infections (including Salmonella osteomyelitis). These infections are especially common in early childhood and can be fatal.
Recurrent ischemia and infarction can cause chronic dysfunction of multiple different organ systems. Complications include ischemic stroke, seizures, avascular necrosis of the hips, renal concentrating defects, renal papillary necrosis, chronic kidney disease, heart failure, pulmonary hypertension, pulmonary fibrosis, and retinopathy.
Heterozygotes
Patients who are heterozygous (Hb AS) do not experience hemolysis or painful crises. However, they do have an increased risk of chronic kidney disease and pulmonary embolism. In addition, rhabdomyolysis and sudden death may occur during sustained, exhausting exercise. Impaired ability to concentrate urine (hyposthenuria) is common. Unilateral hematuria (by unknown mechanisms and usually from the left kidney) can occur but is self-limited. Typical renal papillary necrosis can occur but is less common than among homozygous patients, and there is an association with the extremely rare medullary carcinoma of the kidney.
Symptoms and Signs of Sickle Cell Disease
Most symptoms occur only in patients who are homozygous and result from
Anemia
Vaso-occlusive events resulting in tissue ischemia and infarction
Anemia is usually severe but varies among patients and is usually compensated; mild jaundice and pallor are common.
Hepatosplenomegaly is common in children, but because of repeated infarctions and subsequent fibrosis (autosplenectomy), the spleen in adults is commonly atrophied. Cardiomegaly and systolic ejection (flow) murmurs are common. Cholelithiasis and chronic punched-out skin ulcers around the ankles are common.
Painful vaso-occlusive crisis causes severe pain in long bones, the hands and feet, back, and joints. Hip pain may result from avascular necrosis of the femoral head. Severe abdominal pain, which may be due to hepatic vein thrombosis, may develop with or without vomiting and is usually accompanied by back and joint pain.
Acute chest syndrome is characterized by sudden onset of fever, chest pain, and pulmonary infiltrates. It may follow bacterial pneumonia. Hypoxemia may develop rapidly, causing dyspnea.
Diagnosis of Sickle Cell Disease
DNA testing (prenatal diagnosis)
Peripheral smear
Solubility testing
Hemoglobin electrophoresis (or thin-layer isoelectric focusing)
The type of testing done depends on the age of the patient. DNA testing can be used for prenatal diagnosis or to confirm a diagnosis of the sickle cell genotype. Screening of neonates is available in most US states and involves hemoglobin electrophoresis. Screening and diagnosis in children and adults involve examination of the peripheral smear, hemoglobin solubility testing, and hemoglobin electrophoresis.
Prenatal screening
The sensitivity of prenatal diagnosis has been greatly improved with the availability of PCR (polymerase chain reaction) technology. It is recommended for families at risk for sickle cell (eg, couples with medical or family histories of anemia or of suggestive ethnic background). DNA samples can be obtained by chorionic villus sampling at 10 to 12 weeks' gestation. Amniotic fluid can also be tested at 14 to 16 weeks. Diagnosis is important for genetic counseling.
Newborn screening
Universal testing is currently recommended and is frequently one of a battery of newborn screening tests. To distinguish between hemoglobin (Hb) F, S, A, and C, the recommended tests are hemoglobin electrophoresis using cellulose acetate or acid citrate agar, thin-layer isoelectric focusing, or hemoglobin fractionation by high performance liquid chromatography (HPLC). Repeat testing at age 3 to 6 months may be necessary for confirmation. Solubility testing for Hb S is unreliable during the first few months of life.
Screening and diagnosis of children and adults
Patients with a family history of sickle cell disease or trait should be screened with peripheral smear, hemoglobin (Hb) solubility testing, and hemoglobin electrophoresis.
Patients with symptoms or signs suggesting the disorder or its complications (eg, poor growth, acute and unexplained bone pain, particularly in the fingers, aseptic necrosis of the femoral head, unexplained hematuria), and patients with African ancestry with normocytic anemia (particularly if hemolysis is present) require laboratory tests for hemolytic anemia, hemoglobin electrophoresis, and examination of RBCs for sickling. If sickle cell disease is present, the red blood cell count is usually between 2 and 3 million/microL (2 and 3 x 1012/L) with hemoglobin reduced proportionately; cells are normocytic (microcytosis suggests a concomitant alpha or beta thalassemia). Nucleated RBCs frequently appear in the peripheral blood, and reticulocytosis ≥ 10% is common. Dry-stained smears may show sickled RBCs (crescent-shaped, often with elongated or pointed ends).

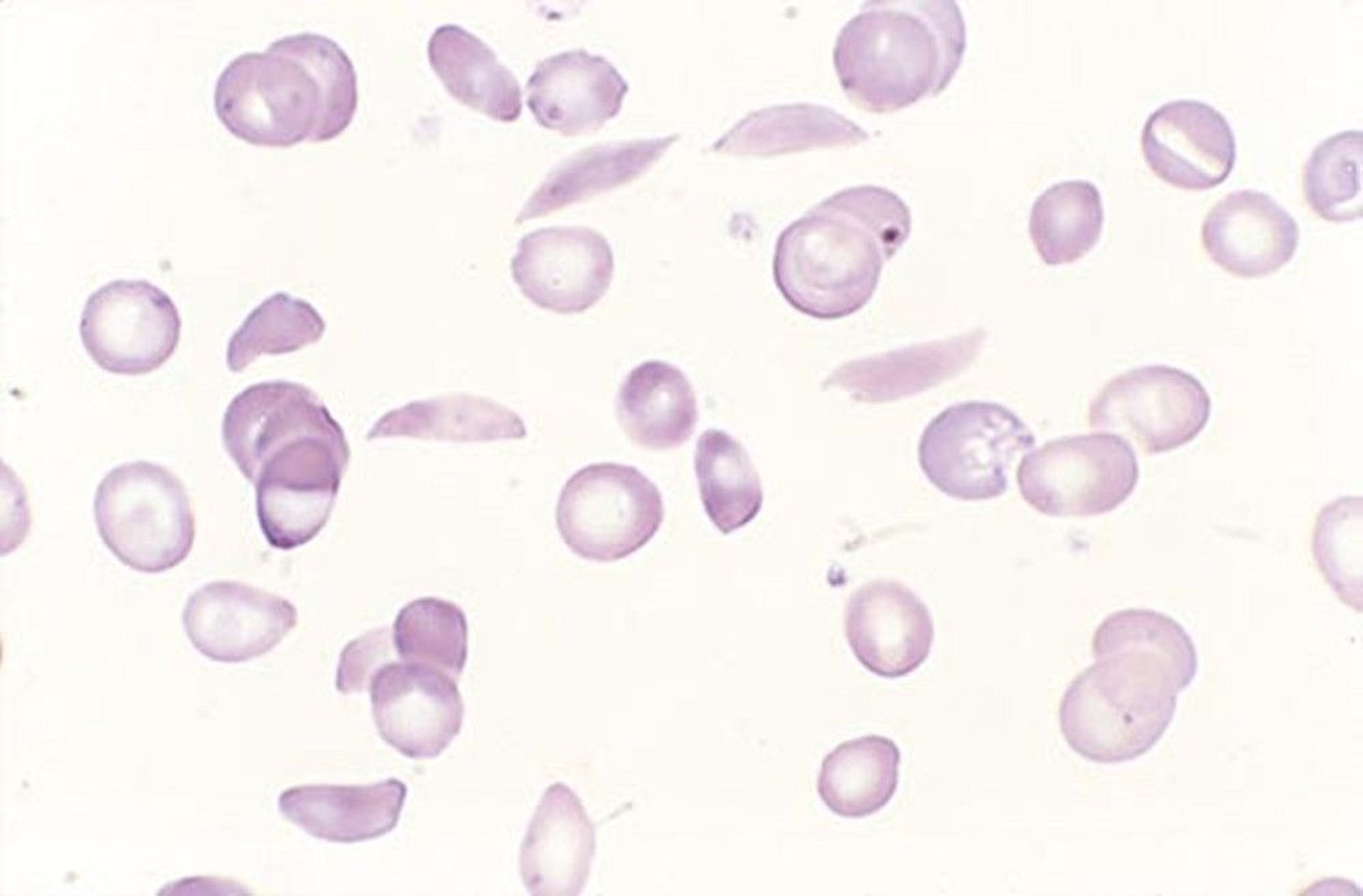
By permission of the publisher. From Tefferi A, Li C. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.
The homozygous state is differentiated from other sickle hemoglobinopathies by electrophoresis showing only Hb S with a variable amount of Hb F. The heterozygote is differentiated by the presence of more Hb A than Hb S on electrophoresis. Hb S must be distinguished from other hemoglobin types with a similar electrophoretic pattern by showing the pathognomonic RBC morphology.
Bone marrow examination is not used for diagnosis. If it is done to differentiate other anemias, it shows hyperplasia, with normoblasts predominating; bone marrow may become aplastic during sickling or severe infections. The erythrocyte sedimentation rate, if done to exclude other disorders (eg, juvenile rheumatoid arthritis causing hand and foot pain), is low.
Incidental findings on skeletal x-rays may include widening of the diploic spaces of the skull and a sun-ray appearance of the diploic trabeculations. The long bones often show cortical thinning, irregular densities, and new bone formation within the medullary canal.
Unexplained hematuria, even among patients not suspected of having sickle cell disease, should prompt consideration of sickle cell trait.
Evaluation of exacerbations
If patients with known sickle cell disease have acute exacerbations, including pain, fever, or other symptoms of infection, aplastic crisis is considered and a complete blood count and reticulocyte count are done. A reticulocyte count < 1% suggests aplastic crisis, particularly when hemoglobin decreases below the patient’s usual level. In a painful crisis without aplasia, the white blood cell count rises, often with a shift to the left, particularly during bacterial infection. The platelet count is usually increased but can fall with the acute chest syndrome. If measured, serum bilirubin is usually elevated (eg, 2 to 4 mg/dL [34 to 68 micromol/L]), and urine may contain urobilinogen.
In patients with chest pain or difficulty breathing, acute chest syndrome and pulmonary embolism are considered; chest x-ray and pulse oximetry are necessary. Because acute chest syndrome is the leading cause of death in sickle cell disease, early recognition and treatment are critical. Hypoxemia or pulmonary parenchymal infiltrates on chest x-ray suggest acute chest syndrome or pneumonia. Hypoxemia without pulmonary infiltrates suggests pulmonary embolism.
In patients with fever, infection and acute chest syndrome are considered; cultures, chest x-ray, and other appropriate diagnostic tests are done.
Prognosis for Sickle Cell Disease
The life span of homozygous patients has steadily increased to > 50 years. Common causes of death are acute chest syndrome, intercurrent infections, pulmonary emboli, infarction of a vital organ, pulmonary hypertension, and chronic kidney disease.
Treatment of Sickle Cell Disease
Broad-spectrum antibiotics (for infection)
Analgesics and IV hydration (for vaso-occlusive pain crisis)
Oxygen (for hypoxia)
Sometimes transfusions
Stem cell transplantation (for advanced complications)
Treatment includes regular health maintenance measures as well as specific treatment of the complications as they arise. Complications are treated supportively. No effective in vivo anti-sickling drug is available. Splenectomy is valueless.
Indications for hospitalization include suspected serious (including systemic) infection, aplastic crisis, acute chest syndrome, and, often, intractable pain or the need for transfusion. Fever alone may not be a reason to hospitalize. However, patients who appear acutely ill and have a temperature > 38° C should be admitted so that cultures can be obtained and IV antibiotics can be given.
Antibiotics
Patients with suspected serious bacterial infections or acute chest syndrome require broad-spectrum antibiotics immediately.
Analgesics
Intravenous hydration
Although dehydration contributes to sickling and may precipitate crises, it is unclear whether vigorous hydration is helpful during crises. Nevertheless, maintaining normal intravascular volume has been a mainstay of therapy.
Oxygen
Oxygen is given if needed to treat hypoxia.
Transfusion
Transfusion is given in many situations in which its efficacy has not been demonstrated. However, chronic transfusion therapy is indicated for prevention of recurrent cerebral thrombosis, especially in children, in an effort to maintain the Hb S percentage less than 30%.
In the acute setting, specific indications for transfusion include
Acute splenic sequestration
Aplastic crises
Cardiopulmonary symptoms or signs (eg, high-output heart failure, hypoxemia with PO2 < 65 mm Hg)
Preoperative use
Priapism
Life-threatening events that would benefit from improved oxygen delivery (eg, sepsis, severe infection, acute chest syndrome, stroke, acute organ ischemia)
Sometimes pregnancy
Transfusion is not helpful during an uncomplicated painful crisis.
Simple transfusion can be done when the goal is to correct anemia, such as during aplastic crisis or splenic or hepatic sequestration. Exchange transfusion is done during severe acute events such as the acute chest syndrome or stroke in order to decrease the Hb S percentage and prevent ischemia. It can be done with modern apheresis machines. If the initial hemoglobin is low (< 7 g/dL [< 70 g/L]), this process cannot be initiated before first transfusing red cells. Partial exchange transfusion minimizes iron accumulation and hyperviscosity.
Curative treatments
Hematopoietic stem cell transplantation remains the only curative treatment for sickle cell disease. Given the risks associated with this therapy, it is generally restricted to patients with advanced disease complications.
Gene therapy or gene editing techniques that reduce the amount of Hb S are currently in clinical trials. This field is rapidly evolving and use of stem cell therapy to treat sickle cell disease will likely expand in the near future.
Health maintenance
For long-term management the following interventions have reduced mortality, particularly during childhood:
Pneumococcal, Haemophilus influenzae influenza (inactivated, not live), and meningococcal vaccines
Early identification and treatment of serious bacterial infections
Prophylactic antibiotics, including continuous prophylaxis with oral penicillin from age 4 months to 6 year
1, 23. While these drugs are currently being incorporated into treatment regimens for sickle cell patients, data on their efficacy remain limited.
Transcranial Doppler flow studies in children can help predict risk of stroke, and many experts recommend annual screening for children from age 2 to 16 years. Children at high risk appear to benefit from prophylactic, chronic exchange transfusions to keep Hb S at < 30% of total hemoglobin; iron overload is common and must be screened for and treated.
For patients receiving frequent red cell transfusions, chelation therapy to prevent or delay complications due to iron overload should be considered.
Treatment references
1. Ataga KI, Kutlar A, Kanter J, et al: Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med 376(5):429–439, 2017. doi: 10.1056/NEJMoa1611770
2. Niihara Y, Miller ST, Kanter J, et al: A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med 379(3):226–235, 2018. doi: 10.1056/NEJMoa1715971
3. Vichinsky E, Hoppe CC, Ataga KI, et al: A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med 381(6):509–519, 2019. doi: 10.1056/NEJMoa1903212
Key Points
Patients homozygous for hemoglobin S have an abnormal beta chain, resulting in fragile, relatively inflexible red blood cells that can plug capillaries, causing tissue infarction and are prone to hemolysis, causing anemia.
Patients have various acute exacerbations including painful crisis, sequestration crisis, aplastic crisis, and acute chest syndrome.
Long-term consequences include pulmonary hypertension, chronic kidney disease, stroke, aseptic necrosis, and increased risk of infection.
Diagnose using hemoglobin electrophoresis.
For acute crises, give opioid analgesics for pain, check for worsening anemia (suggesting aplastic or sequestration crisis) and signs of acute chest syndrome or infection, restore normal intravascular volume using 0.9% saline and then give maintenance fluids.
More Information
The following is an English-language resource that may be useful. Please note that THE MANUAL is not responsible for the content of this resource.
Sickle Cell Disease Association of America: provides comprehensive patient education and support, including peer mentoring, to patients with sickle cell disease






