


Platelets are circulating cell fragments that function in the clotting system. Thrombopoietin helps control the number of circulating platelets by stimulating the bone marrow to produce megakaryocytes, which in turn shed platelets from their cytoplasm. Thrombopoietin is produced in the liver at a constant rate and its circulating level is determined by how much is bound to circulating platelets and possibly to bone marrow megakaryocytes and the extent to which circulating platelets are cleared. Platelets circulate for 7 to 10 days. About one third are always transiently sequestered in the spleen.
The platelet count is normally 140,000to 440,000/mcL (140 to 440 × 109/L). However, the count can vary slightly according to menstrual cycle phase, decrease during near-term pregnancy (gestational thrombocytopenia), and increase in response to inflammatory cytokines (secondary, or reactive, thrombocytosis). Platelets are eventually destroyed by apoptosis, a process independent of the spleen.
Platelet disorders include
An abnormal increase in platelets (thrombocythemia and reactive thrombocytosis)
A decrease in platelets (thrombocytopenia)
Platelet dysfunction
Any of these conditions, even those in which platelets are markedly increased, may cause defective formation of hemostatic plugs and bleeding.
The risk of bleeding is inversely proportional to the platelet count and platelet function (see table Platelet Count and Bleeding Risk
Etiology of Platelet Disorders
Thrombocythemia and thrombocytosis
Essential thrombocythemia is a myeloproliferative neoplasm (previously called a myeloproliferative disorder) involving overproduction of platelets because of a clonal abnormality of a hematopoietic stem cell. There is no correlation between the platelet count and risk of thrombosis, but some patients with extreme thrombocytosis (ie, > 1,000,000/mcL [> 1000 × 109von Willebrand disease).
Reactive thrombocytosis is platelet overproduction in response to another disorder. There are many causes, including acute infection, chronic inflammatory disorders (eg, rheumatoid arthritis, inflammatory bowel disease, tuberculosis, sarcoidosis), iron deficiency, and certain cancers. Reactive thrombocytosis is not typically associated with an increased risk of thrombosis or bleeding.
Thrombocytopenia
Causes of thrombocytopenia can be classified by mechanism (see table Classification of Thrombocytopenia) and include
Decreased platelet production
Increased splenic sequestration of platelets with normal platelet survival
Increased platelet destruction or consumption (both immunologic and nonimmunologic causes)
Dilution of platelets
A large number of drugs may cause thrombocytopenia, typically by triggering immunologic destruction.
Overall, the most common specific causes of thrombocytopenia include
Pregnancy (gestational thrombocytopenia; HELLP syndrome [hemolysis, elevated liver enzymes, and low platelets])
Systemic infection
Immune disorders (eg, immune thrombocytopenia [ITP], antiphospholipid antibody syndrome, systemic lupus erythematosus)
Platelet dysfunction
Platelet dysfunction may stem from an intrinsic platelet defect or from an extrinsic factor that alters the function of normal platelets. Dysfunction may be hereditary or acquired. Hereditary disorders of platelet function consist of von Willebrand disease, the most common hereditary hemorrhagic disease, and hereditary intrinsic platelet disorders, which are much less common. Acquired disorders of platelet dysfunction
Symptoms and Signs of Platelet Disorders
Platelet disorders result in a typical pattern of bleeding:
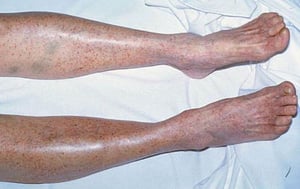
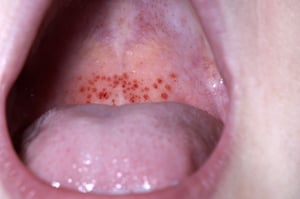
Multiple petechiae in the skin (typically most evident on the lower legs)
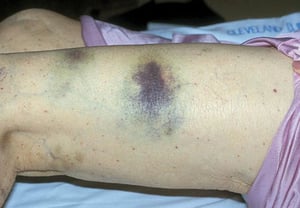
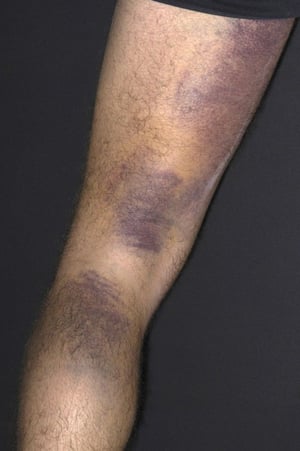
Scattered small ecchymoses at sites of minor trauma or venipuncture sites
Mucosal bleeding (oropharyngeal, nasal, gastrointestinal, genitourinary)
Excessive bleeding after surgery
Extensive menstrual bleeding
By permission of the publisher. From Deitcher S. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.
By permission of the publisher. From Deitcher S. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
DR P. MARAZZI/SCIENCE PHOTO LIBRARY

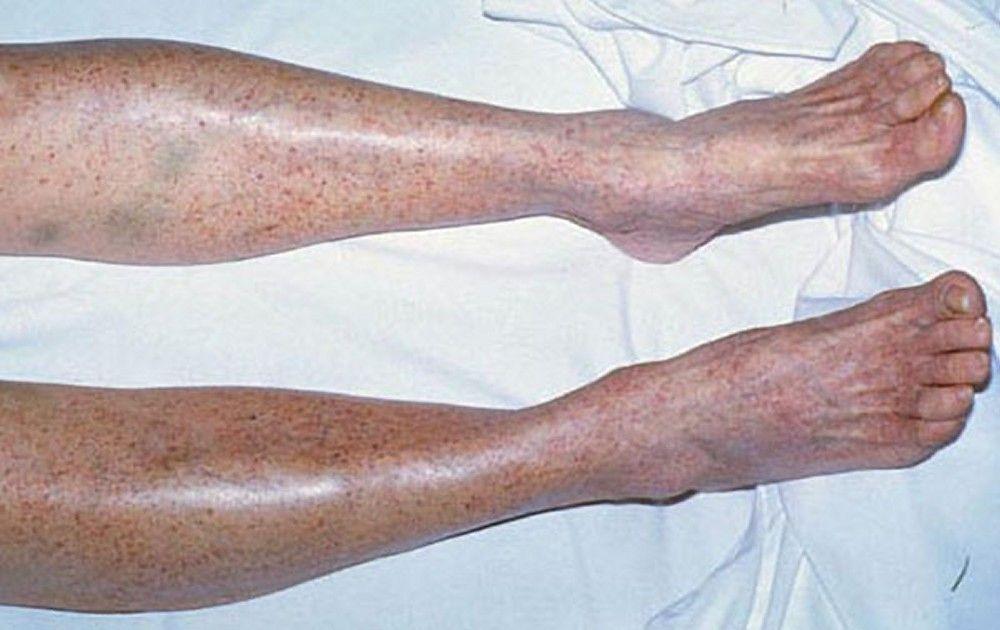
By permission of the publisher. From Deitcher S. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.

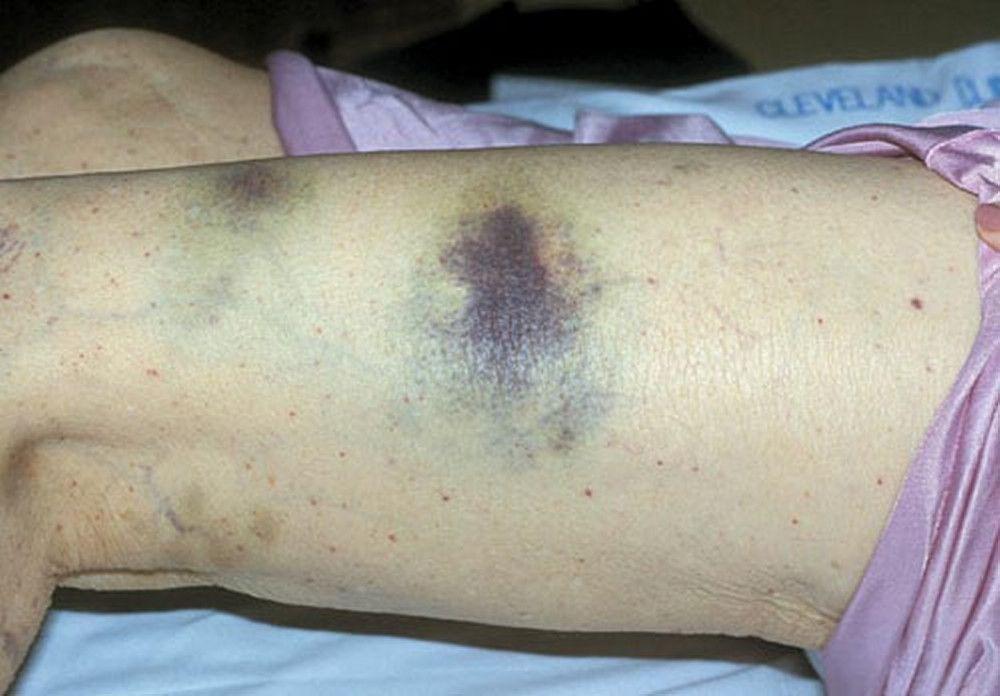
By permission of the publisher. From Deitcher S. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.

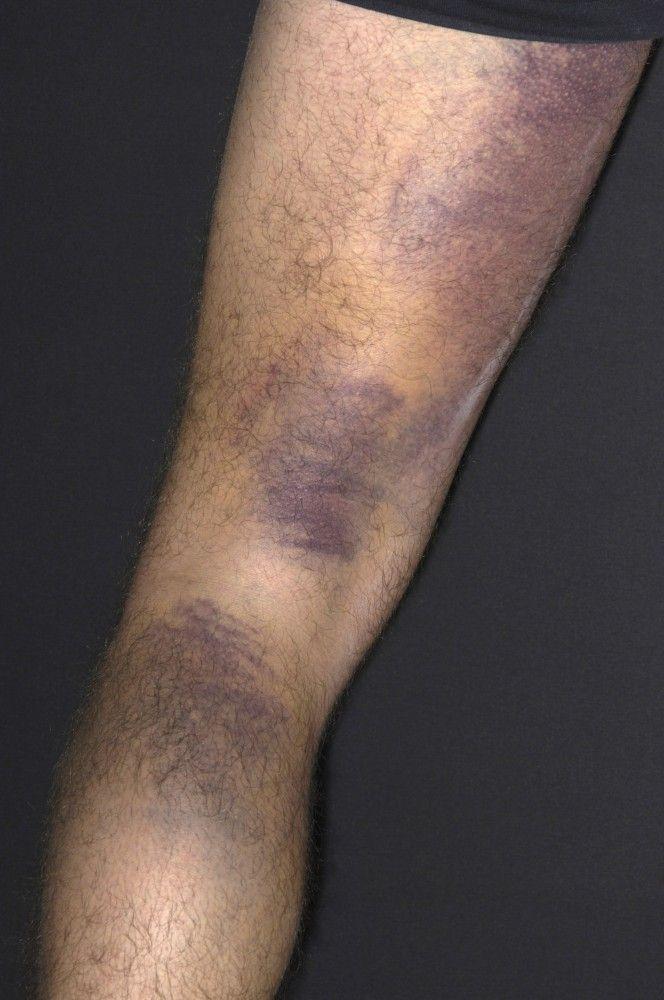
DR P. MARAZZI/SCIENCE PHOTO LIBRARY

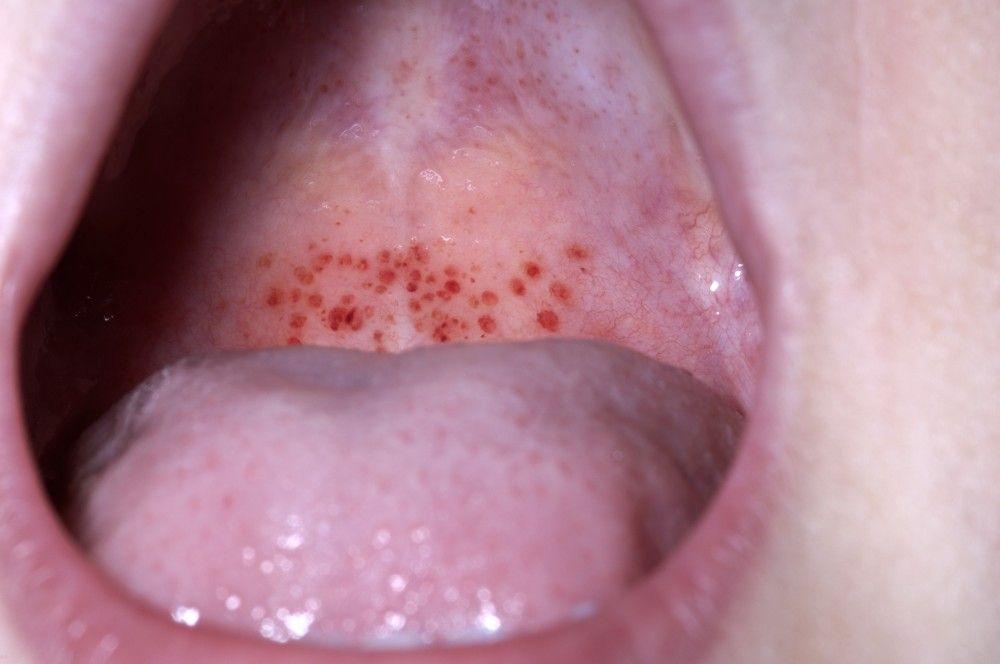
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
Heavy gastrointestinal bleeding and bleeding into the central nervous system are rare but may be life threatening. However, bleeding into tissues (eg, deep visceral hematomas or hemarthroses) rarely occurs with thrombocytopenia; instead, patients usually have immediate and superficial bleeding following an injury. Bleeding into the tissues (often delayed for up to a day after trauma) suggests a coagulation disorder (eg, hemophilia).
Diagnosis of Platelet Disorders
Clinical presentation of petechiae and mucosal bleeding
Complete blood count (CBC) with platelets, coagulation studies, peripheral blood smear
Sometimes bone marrow aspiration
Sometimes von Willebrand antigen, platelet-binding activity, and multimer studies
Platelet disorders are suspected in patients with petechiae and mucosal bleeding. A CBC with platelet count, coagulation studies, and a peripheral blood smear are obtained. Excessive platelets and thrombocytopenia are diagnosed based on the platelet count. Coagulation studies are normal unless there is a simultaneous coagulopathy. In patients with a normal CBC, platelet count, international normalized ratio (INR), and partial thromboplastin time (PTT), platelet or vessel wall dysfunction is suspected.
Pearls & Pitfalls
|
Thrombocytopenia
Peripheral smear examination is important in patients with thrombocytopenia because automated platelet counts sometimes show pseudothrombocytopenia due to platelet clumping caused by the ethylenediaminetetraacetic acid (EDTA) reagent present in some blood collection tubes. Also, schistocytes may be seen, which can indicate valvular hemolysis, thrombotic thrombocytopenic purpura (TTP), hemolytic-uremic syndrome (HUS), or disseminated intravascular coagulation (DIC—see table Peripheral Blood Findings in Thrombocytopenic Disorders).
Bone marrow aspiration is often indicated if the smear shows abnormalities other than thrombocytopenia, such as nucleated red blood cells (RBCs) or abnormal or immature white blood cells (WBCs). Bone marrow aspiration reveals the number and appearance of megakaryocytes and is the definitive test for many disorders that cause bone marrow failure. If the bone marrow is normal but the spleen is enlarged, increased splenic sequestration is the likely cause of thrombocytopenia. If the bone marrow is normal and the spleen is not enlarged, excess platelet destruction is the likely cause.
However, normal number and appearance of megakaryocytes does not always indicate normal platelet production. For example, in many patients with immune thrombocytopenia (ITP), platelet production may be decreased despite the normal appearance and increased number of megakaryocytes. In fact, bone marrow examination is rarely required in patients who present with typical features of immune thrombocytopenia.
The immature platelet fraction in peripheral blood is sometimes a useful measure in patients with thrombocytopenia, since it is elevated when the bone marrow is producing platelets and not increased when marrow platelet production is reduced, similar to the reticulocyte count in anemia.
Measurement of antiplatelet antibodies may be clinically useful in some patients to distinguish ITP from other causes of thrombocytopenia (1). HIV testing is done in patients with or at risk of HIV infection, hepatitis B or C infection, or HIV and hepatitis coinfection.

By permission of the publisher. From Tefferi A, Li C. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.
Suspected platelet dysfunction
In patients with suspected hereditary dysfunction, platelet aggregation tests may identify a defect in how the platelet responds to various platelet agonists (adenosine
Platelet dysfunction caused by systemic disorders is typically mild and of minor clinical importance. In these patients, the causative systemic disorder is the clinical concern, and hematologic tests are unnecessary.
Diagnosis reference
1. Al-Samkari H, Rosovsky RP, Karp Leaf RS: A modern reassessment of glycoprotein-specific direct platelet autoantibody testing in immune thrombocytopenia. Blood Adv 4(1):9–18, 2020. doi: 10.1182/bloodadvances.2019000868
Treatment of Platelet Disorders
Stopping drugs that impair platelet function
Rarely platelet transfusions
Rarely antifibrinolytic drugs
Patients may require platelet transfusion, but transfusions are given only in limited situations. Prophylactic transfusions are used sparingly because they may lose their effectiveness with repeated use due to the development of platelet alloantibodies.
Active bleeding
Severe thrombocytopenia (eg, platelet count < 10,000/mcL [< 10 × 109/L)
A need for an invasive procedure
In thrombocytopenia caused by platelet destruction, transfusions are reserved for life-threatening,central nervous system or ocular bleeding.






