


Hemostasis, the arrest of bleeding from an injured blood vessel, requires the combined activity of
Vascular factors
Platelets
Plasma coagulation factors
Regulatory mechanisms counterbalance the tendency of clots to form. Hemostatic abnormalities can lead to excessive bleeding or thrombosis.
Vascular Factors of Hemostasis
Vascular factors reduce blood loss due to trauma through local vasoconstriction (an immediate reaction to injury) and compression of injured vessels by extravasation of blood into surrounding tissues. Vessel wall injury triggers the attachment and activation of platelets and the generation of fibrin polymers from fibrinogen; platelets and fibrin combine to form a clot.
Platelets
Various mechanisms, including endothelial cell nitric oxide and prostacyclin, promote blood fluidity by preventing platelet aggregation and dilating intact blood vessels. These mediators are no longer produced when the vascular endothelium is disrupted. Under these conditions, platelets adhere to the damaged intima and form aggregates. Initial platelet adhesion is to long strings of von Willebrand factor (VWF) that have been previously secreted by, and anchored to, stimulated endothelial cells. VWF binds to receptors on the platelet surface membrane (glycoprotein Ib/IX). Platelets anchored to the vessel wall undergo activation. During activation, platelets release mediators of aggregation, including adenosine diphosphate (ADP) from storage granules.
Other biochemical changes resulting from activation include
Hydrolysis of membrane phospholipids
Inhibition of adenylate cyclase
Mobilization of intracellular calcium
Phosphorylation of intracellular proteins
Arachidonic acid is converted to thromboxane A2; this reaction requires platelet cyclooxygenase and is inhibited irreversibly by aspirin and reversibly by many NSAIDs (nonsteroidal anti-inflammatory drugs).
ADP, thromboxane A2, and other mediators induce activation and aggregation of additional platelets on the injured endothelium. Platelet receptors for ADP include the P2Y12 receptor, which sends signals to suppress adenylate cyclase, decreases cyclic adenosine monophosphate (cAMP) levels, and promotes activation of the glycoprotein IIb/IIIa receptor (assembled on the activated platelet surface membrane from glycoproteins IIb and IIIa). Fibrinogen binds to the glycoprotein IIb/IIIa complexes of adjacent platelets, connecting them into aggregates.
Platelets provide surfaces for the assembly and activation of coagulation complexes and the generation of thrombin. Thrombin converts fibrinogen into fibrin monomers, and the fibrin monomers polymerize into fibrin polymers that bind aggregated platelets into platelet-fibrin hemostatic plugs.
Plasma Coagulation Factors
Coagulation factors interact on platelet and endothelial cell surfaces to produce thrombin, which converts fibrinogen to fibrin. By radiating from and anchoring the hemostatic plug, fibrin strengthens the clot.
In the intrinsic pathway, factor XII, high molecular weight kininogen, prekallikrein, and activated factor XI (factor XIa) interact to produce factor IXa from factor IX. Factor IXa then combines with factor VIIIa and procoagulant phospholipid (present on the surface of activated platelets, endothelial cells, and tissue cells to form a complex that activates factor X.
In the extrinsic pathway,factor VIIa and tissue factor (TF) directly activate factor X (and, perhaps, also factor IX—see figure Pathways in Blood Coagulation and table Components of Blood Coagulation Reactions).
The coagulation factors are produced in the liver with the exception of factor VIII, which is synthesized in liver sinusoidal cells and endothelial cells outside the liver. Tissue factor expression is normally restricted to perivascular cells so that extrinsic pathway activation only occurs in the event of vessel wall injury.
Pathways in Blood Coagulation

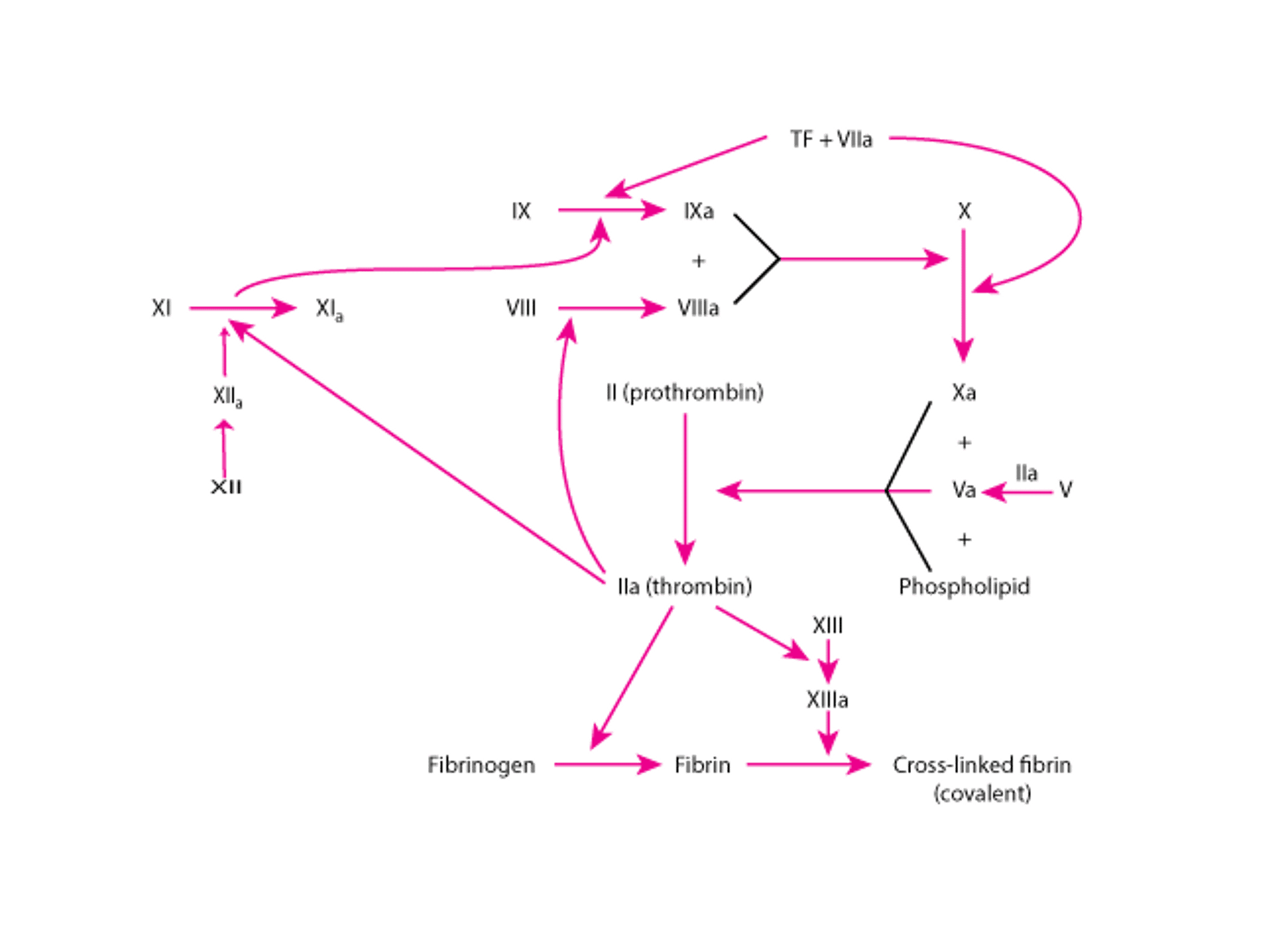
Activation of the intrinsic or extrinsic pathway activates the common pathway, resulting in formation of the fibrin clot. Three steps are involved in common pathway activation:
Prothrombinase is generated on the surface of activated platelets, endothelial cells, and tissue cells. Prothrombinase is a complex of an enzyme, factor Xa, and a cofactor, factor Va, on a procoagulant phospholipid surface.
Prothrombinase cleaves prothrombin to thrombin.
Thrombin induces the generation of fibrin monomers and polymers from fibrinogen and activates soluble factors V, VIII, and XI. Thrombin also activates factor XIII, an enzyme that catalyzes formation of stronger, covalent bonds between adjacent fibrin monomers.
Calcium ions are required in most thrombin-generating reactions and, therefore, calcium-chelating agents (eg, citrate, ethylenediaminetetraacetic acid) are used in vitro as anticoagulants. Vitamin K–dependent clotting factors (factors II, VII, IX, and X) normally bind to phospholipid surfaces through calcium bridges to function in blood coagulation. Coagulation reactions cannot occur properly in the absence of vitamin K. Vitamin K-dependent coagulation regulatory proteins include protein C and protein S.
Although the coagulation pathways are helpful in understanding mechanisms and laboratory evaluation of coagulation disorders, in vivo coagulation does not include factor XII, prekallikrein, or high molecular weight kininogen. People with hereditary deficiencies of these factors have no bleeding abnormality. People with hereditary factor XI deficiency may have a mild to moderate bleeding disorder. In vitro, soluble factor XI can be activated by thrombin. There is, however, no consistent relationship between plasma factor XI levels and the likelihood or extent of bleeding. Soluble factor IX can be activated in vitro both by factor XIa and factor VIIa/tissue factor complexes.
In vivo, initiation of the extrinsic pathway occurs when injury to blood vessels brings blood into contact with tissue factor on membranes of cells within and around the vessel walls. This contact with tissue factor generates factor VIIa/tissue factor complexes that activate factor X (and possibly factor IX). Factor IXa, combined with its cofactor, factor VIIIa, on phospholipid membrane surfaces also generates factor Xa. Factor X activation by factor IXa/VIIIa complexes is required for normal hemostasis. This requirement for factors VIII and IX explains why hemophilia type A (deficiency of factor VIII) or type B (deficiency of factor IX) results in bleeding. Factor X activation by factor VIIa/tissue factor complexes in the extrinsic coagulation pathway does not generate sufficient thrombin (and fibrin) to prevent bleeding in patients with severe hemophilia A or B.
Regulation of Coagulation
Several inhibitory mechanisms prevent activated coagulation reactions from amplifying uncontrollably, causing extensive local thrombosis or disseminated intravascular coagulation. These mechanisms include
Inactivation of coagulation factors
Fibrinolysis
Hepatic clearance of activated clotting factors
Inactivation of coagulation factors
Plasma protease inhibitors (antithrombin, tissue factor pathway inhibitor, alpha 2-macroglobulin, and heparin cofactor II) inactivate coagulation enzymes. Antithrombin inhibits thrombin, factor Xa, factor XIa, and factor IXa.
Two vitamin K–dependent proteins, protein C and free protein S, form a complex that inactivates factors VIIIa and Va by proteolysis. Thrombin, when bound to a receptor on endothelial cells (thrombomodulin [CD141]), activates protein C. Activated protein C, in combination with free protein S and an endothelial cell protein C receptor, proteolyzes and inactivates factors VIIIa and Va.
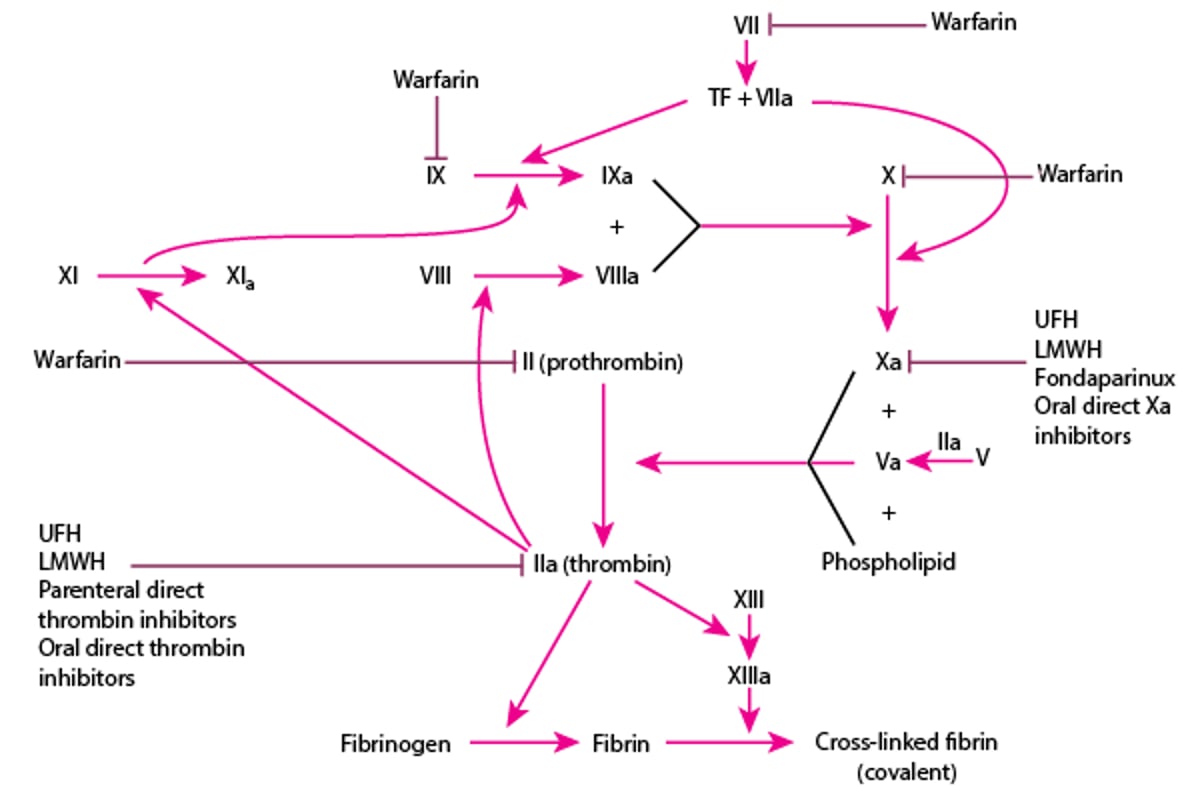
In addition to normally present inactivators, there are a number of anticoagulant medications that potentiate the inactivation of coagulation factors (see figure Anticoagulants and Their Sites of Action).
Heparin enhances antithrombin activity. Unfractionated heparin (UFH) and low molecular weight heparins (LMWH) enhance activity of antithrombin to inactivate factors IIa (thrombin
heparin structure, enhances antithrombin inactivation of factor Xa but not factor IIa (thrombin).
Parenteral direct thrombin
The direct oral anticoagulants include the thrombinfactor Xaatrial fibrillation, deep venous thrombosis (DVT), and pulmonary embolism (PE).
Anticoagulants and Their Sites of Action
LMWH = low molecular weight heparin; TF = tissue factor; UFH = unfractionated heparin. |

Fibrinolysis
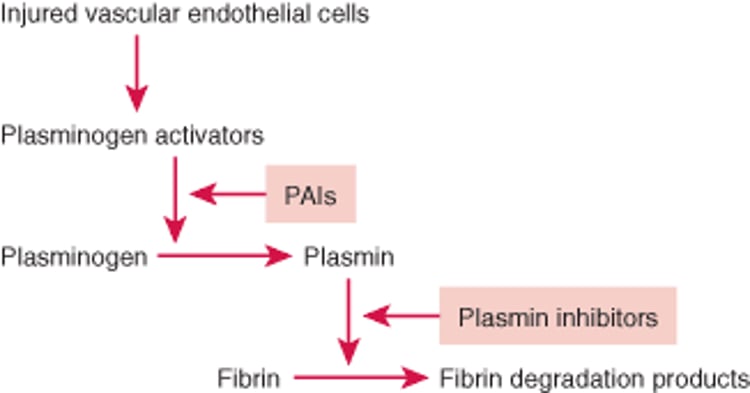
Fibrin deposition and lysis are normally balanced to maintain temporarily, and subsequently remove, the hemostatic seal during repair of an injured vessel wall. The fibrinolytic system dissolves fibrin by means of plasmin, a proteolytic enzyme. Fibrinolysis is activated by plasminogen activators released from vascular endothelial cells. Plasminogen activators and plasminogen (from plasma) bind to fibrin, and plasminogen activators cleave plasminogen into plasmin (see figure Fibrinolytic Pathway). Plasmin then proteolyzes fibrin into soluble fibrin degradation products that are swept away in the circulation and metabolized by the liver.
Fibrinolytic Pathway
Fibrin deposition and fibrinolysis must be balanced during repair of an injured blood vessel wall. Injured vascular endothelial cells release plasminogen activators (tissue plasminogen activator, urokinase), activating fibrinolysis. Plasminogen activators cleave plasminogen into plasmin, which dissolves clots. Fibrinolysis is controlled by plasminogen activator inhibitors (PAIs; eg, PAI-1) and plasmin inhibitors (eg, alpha 2-antiplasmin). |

There are several plasminogen activators:
Tissue plasminogen activator (tPA), from endothelial cells, is a poor activator of plasminogen when free in solution but an efficient activator when bound to fibrin in proximity to plasminogen.
Urokinase exists in single-chain and double-chain forms with different functional properties. Single-chain urokinase cannot activate free plasminogen but, like tPA, can readily activate plasminogen bound to fibrin. A trace concentration of plasmin cleaves single-chain to double-chain urokinase, which activates plasminogen in solution, as well as plasminogen bound to fibrin. Epithelial cells that line excretory passages (eg, renal tubules, mammary ducts) release urokinase, which is the physiologic activator of fibrinolysis in these channels.
Streptokinase, a bacterial product not normally found in the body, is another potent plasminogen activator.
Regulation of fibrinolysis
Fibrinolysis is regulated by plasminogen activator inhibitors (PAIs) and plasmin inhibitors that slow fibrinolysis. PAI-1, the most important PAI, inactivates tPA, and urokinase and is released from vascular endothelial cells and activated platelets. The primary plasmin inhibitor is alpha 2-antiplasmin, which quickly inactivates any free plasmin escaping from clots. Some alpha 2-antiplasmin is also cross-linked to fibrin polymers by the action of factor XIIIa during clotting. This cross-linking may prevent excessive plasmin activity within clots.
Tissue plasminogen activator and urokinase are rapidly cleared by the liver, which is another mechanism of preventing excessive fibrinolysis.






