


Mixed connective tissue disease is an uncommon, specifically defined syndrome characterized by clinical features of systemic lupus erythematosus, systemic sclerosis, and polymyositis with very high titers of circulating antinuclear antibody to a ribonucleoprotein antigen. Hand swelling, Raynaud syndrome, polyarthralgia, inflammatory myopathy, esophageal hypomotility, and interstitial lung disease are common. Diagnosis is by the combination of clinical features, antibodies to ribonucleoprotein, and absence of antibodies specific for other autoimmune diseases. Treatment varies with disease severity and organ involvement but usually includes corticosteroids and additional immunosuppressants.
Mixed connective tissue disease (MCTD) occurs worldwide and in all races, with a peak incidence in adolescence and the 20s. About 80% of people who have this disease are women. The cause of MCTD is unknown. In many patients, the disorder evolves into classic systemic sclerosis or systemic lupus erythematosus (SLE).
Symptoms and Signs of MCTD
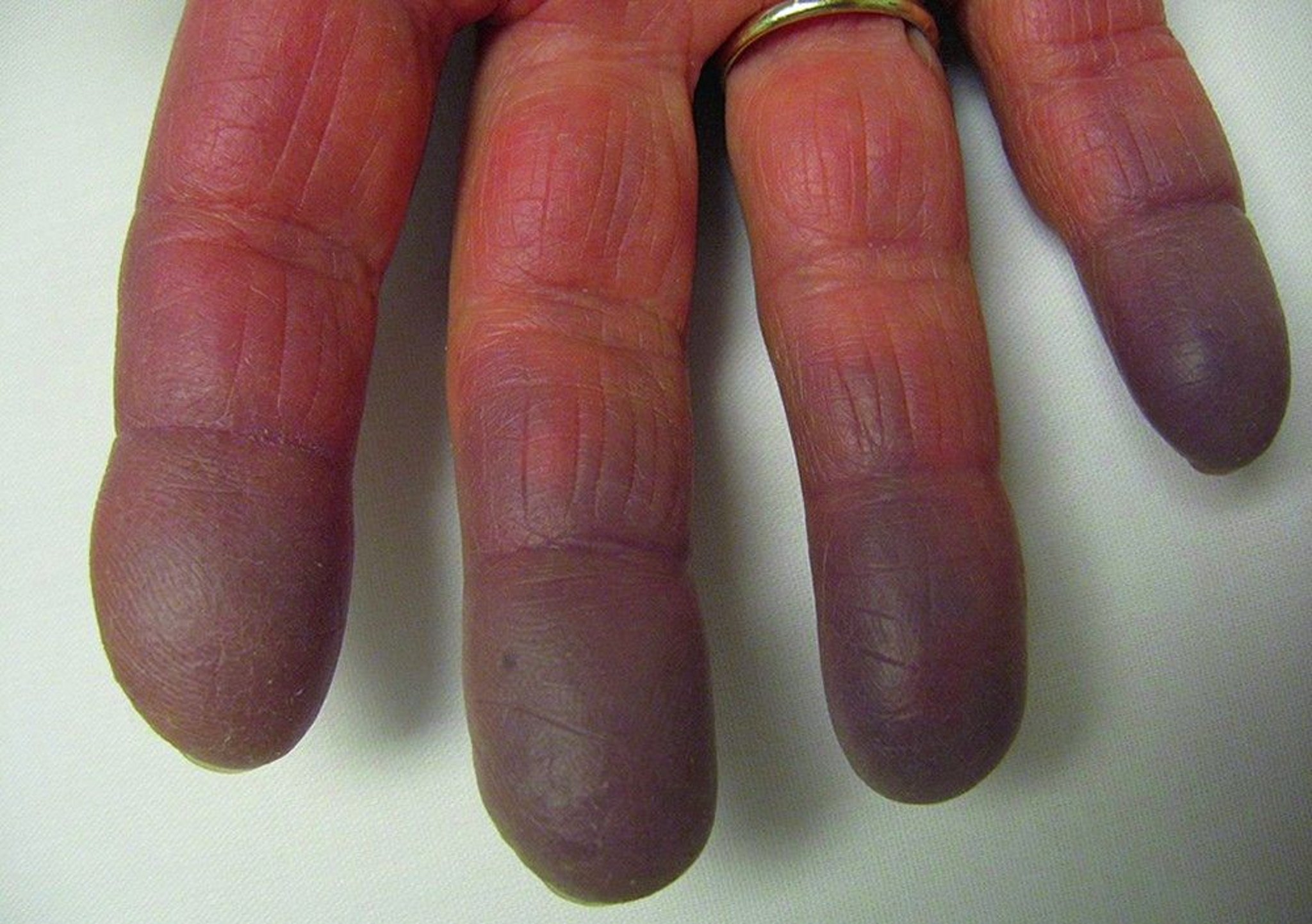
Raynaud syndrome (Raynaud phenomenon) may precede other manifestations by years. Frequently, the first manifestations resemble early SLE, systemic sclerosis, polymyositis, or even rheumatoid arthritis. Many patients appear to have an undifferentiated connective tissue disease initially. The disease manifestations may progress and become widespread, and the clinical pattern changes over time.
Initial, diffuse swelling of the hands is typical but not universal. Skin findings include lupus or dermatomyositis-like rashes. Diffuse systemic sclerosis–like skin changes and ischemic necrosis or ulceration of the fingertips may develop.


© Springer Science+Business Media

© Springer Science+Business Media
Almost all patients have polyarthralgias, and 75% have frank arthritis. Often the arthritis is nondeforming, but erosive changes and deformities similar to those in rheumatoid arthritis (eg, boutonnière and swan-neck deformities) may be present. Proximal muscle weakness with or without tenderness is common, typically among people who have elevated levels of muscle enzymes (eg, creatine kinase).
Renal involvement (most commonly membranous nephropathy) occurs in about 25% of patients and is typically mild; severe involvement, with morbidity or mortality, is atypical for MCTD. The lungs are affected in up to 75% of patients with MCTD. Interstitial lung disease is the most common lung manifestation; pulmonary hypertension is a major cause of death. Heart failure can occur. Sjögren syndrome may develop.
Diagnosis of MCTD
Testing for antinuclear antibodies (ANA), antibodies to extractable nuclear antigen (antibodies to U1 ribonucleoprotein, or RNP), and Smith (Sm) and anti-DNA antibodies
Organ involvement determined as clinically indicated
MCTD should be suspected when additional overlapping features are present in patients appearing to have systemic lupus erythematosus (SLE), systemic sclerosis, or polymyositis.
Tests for ANA and antibody to U1 RNP antigen are done first. Almost all patients have high titers of fluorescent ANA that produce a speckled pattern. Antibodies to U1 RNP are present, usually at very high titers. Antibodies to the ribonuclease-resistant Sm component of extractable nuclear antigen (anti-Sm antibodies) and to double-stranded DNA (negative in MCTD by definition) are measured to exclude other disorders. The presence of anti-RNP antibodies is not sufficient to make the diagnosis of MCTD; typical clinical findings are also required.
Rheumatoid factors are frequently present, and titers may be high. The erythrocyte sedimentation rate is frequently elevated.
Pulmonary hypertension should be detected as early as possible with pulmonary function testing and echocardiography. Further evaluation depends on symptoms and signs; manifestations of myositis, renal involvement, or pulmonary involvement prompt tests of those organs.
Creatine kinase, MRI, electromyogram, or muscle biopsy can help confirm the presence of myositis as part of MCTD.
Prognosis for MCTD
The overall 10-year survival rate is about 80%, but prognosis depends largely on which manifestations predominate. Patients with features of systemic sclerosis and polymyositis have a worse prognosis. Patients are at increased risk of atherosclerosis. Causes of death include pulmonary hypertension, renal failure, myocardial infarction, colonic perforation, disseminated infection, and cerebral hemorrhage. Some patients have sustained remissions for many years without treatment.
Treatment of MCTD
General management and initial drug therapy are tailored to the specific clinical problem and are similar to those of SLEmyositis or systemic sclerosis, treatment is as for those diseases.
Patients with Raynaud syndrome
All patients should be closely monitored for atherosclerosis. Patients on long-term corticosteroid therapy should receive osteoporosis prophylaxis. If combination immunosuppressive therapy is used, patients should be given prophylaxis for opportunistic infections, such as Pneumocystis jirovecii (see prevention of Pneumocystis jirovecii pneumonia), and vaccines against common infections (eg, streptococcal pneumonia, influenza, COVID-19).
Some experts encourage screening for pulmonary hypertension on a periodic basis with pulmonary function testing and/or echocardiography every 1 to 2 years, depending on symptoms.
Key Points
MCTD most often resembles SLE, systemic sclerosis, and/or polymyositis.
Typically, ANA and antibodies to U1 RNP are present and anti-Sm and anti-DNA antibodies are absent, but the presence of anti-RNP antibodies is not sufficient to make the diagnosis.
Anticipate pulmonary hypertension.
Treat mild disease with NSAIDs or antimalarials and more severe disease with corticosteroids and other immunosuppressants.






