Benign bone tumors include benign giant cell tumors of bone, chondroblastomas, chondromyxoid fibromas, enchondromas, nonossifying fibromas, osteoblastomas, osteochondromas, and osteoid osteomas.
Benign cysts include aneurysmal bone cysts and unicameral bone cysts.
Fibrous dysplasia can also affect bones.
(See also Overview of Bone and Joint Tumors.)
Aneurysmal bone cyst
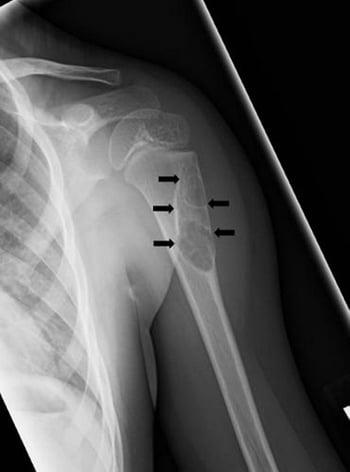
An aneurysmal bone cyst is an idiopathic expansile lesion that usually develops before age 25 years. This cystic lesion usually occurs in the metaphyseal region of the long bones, but almost any bone may be affected. It tends to grow slowly. A periosteal new bone shell forms around the expansile lesion and is often wider than the original bone. Pain and swelling are common. The lesion may be present for a few weeks to a year before diagnosis.
The appearance on x-ray is often characteristic: The lucent area is usually well circumscribed and eccentric; the periosteum bulges (balloons), extending into the soft tissues, and may be surrounded by new bone formation. MRI typically shows fluid-fluid levels. On imaging, some aneurysmal bone cyst–like lesions may appear more ominous, having characteristics similar to osteosarcoma, and thus should raise suspicion of telangiectatic osteosarcoma. A solid variant aneurysmal bone cyst can be confused radiographically for a giant cell tumor of bone at the very end of the bone.

Benign giant cell tumor of bone
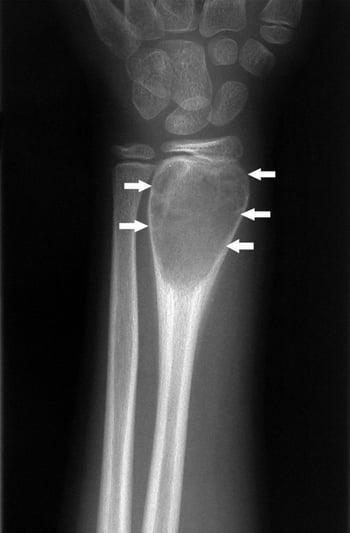
Benign giant cell tumors of bone, which most commonly affect people in their 20s and 30s, occur in the epiphyseal and distal metaphyseal-epiphyseal areas. These tumors are considered locally aggressive. They continue to enlarge and destroy bone and may eventually erode the rest of the bone and extend into the soft tissues. They may cause pain. These tumors are notorious for their tendency to recur. Rarely, a giant cell tumor of bone may metastasize to the lung, even though it remains histologically benign.
Benign giant cell tumors of bone appear as expansile lytic lesions on imaging. On imaging studies, there is a margin without a sclerotic rim where the tumor ends and normal trabecular bone begins. Biopsy is necessary. Because a giant cell tumor of bone may metastasize to the lung, a chest CT is done as part of initial staging.


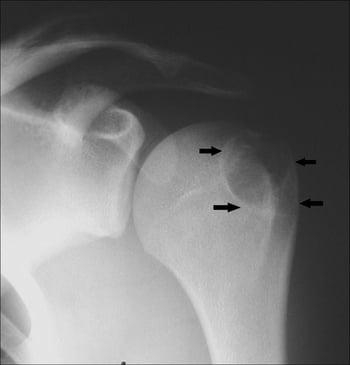
Chondroblastoma
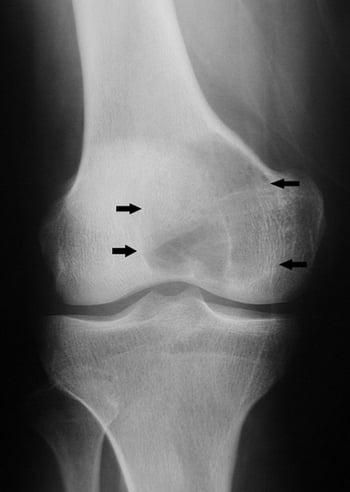
Chondroblastoma is rare and occurs most commonly among people aged 10 to 20. Arising in the epiphysis, this tumor may continue to grow and destroy bone and the joint.
It appears on imaging studies as a sclerotic marginated cyst containing spots of punctate calcification. MRI can help diagnostically by showing significant edema around the lesion.
The tumor must be surgically removed by curettage, and the cavity must be bone grafted. Local recurrence rate is about 10 to 20%, and recurrent lesions often resolve with repeat bone curettage and bone grafting.

Chondromyxoid fibroma
Chondromyxoid fibroma is very rare and usually occurs before age 30.
The appearance on imaging studies, which is usually eccentric, sharply circumscribed, lytic, and located near the end of long bones, suggests the diagnosis of chondromyxoid fibroma. The proximal tibia and iliac wing are typical locations.

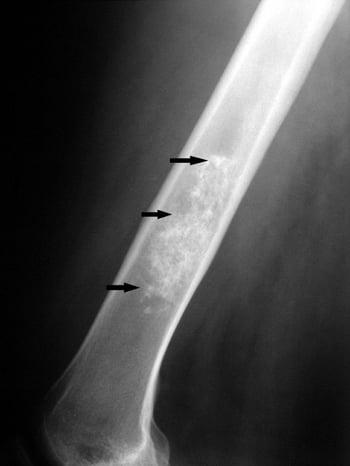
Enchondroma
Enchondromas may occur at any age but tend to manifest in people aged 10 to 40. They are usually located within the medullary bone metaphyseal-diaphyseal region. These tumors are usually asymptomatic but may enlarge and become painful. They are often found when x-rays are taken for another reason. Periosteal chondromas are similar cartilage lesions that occur on the surface of bones.
An asymptomatic enchondroma does not need biopsy, excision, or other treatment (usually curettage); however, follow-up imaging studies are indicated to rule out the rare disease progression to chondrosarcoma. These studies are done at 6 months and again at 1 year or whenever symptoms develop.
Patients with multiple enchondromas (Ollier disease) and especially multiple enchondromatosis with soft tissue hemangiomas (Maffucci syndrome) have a much higher risk of chondrosarcoma.

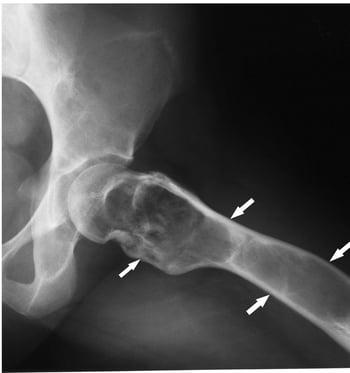
Fibrous dysplasia
Fibrous dysplasia involves abnormal bone development during childhood. It weakens bone. Fibrous dysplasia may affect one or several bones. Multiple fibrous dysplasias, cutaneous pigmentation, and endocrine abnormalities may be present (Albright syndrome or McCune-Albright syndrome). The abnormal bone lesions of fibrous dysplasia commonly stop developing at puberty. They rarely undergo malignant degeneration.
On x-ray, the lesions can appear cystic and may be extensive and deforming. On imaging, the lesions have a classic ground-glass appearance. Proximal femur lesions, because the bone is weakened, undergo plastic deformation, resulting in the appearance of a "shepherd's crook" deformity on x-ray.

Melorheostosis
Melorheostosis is a rare developmental disease of cortical bone. It is a mesenchymal dysplasia of the cortex that may be linked to a random MAP2K1 gene mutation. The process starts in childhood with thickening of the bone cortex on both the outer surface (periosteal) and inner surface (endosteal). The process proceeds very slowly in the cortex. Melorheostosis usually affects only a single bone (monostotic) but may affect other bones (polyostotic) of the same limb (somatic distribution). The tibia and femur are common sites.
Patients often present with discomfort, and x-rays demonstrate thickening of the cortex of the involved bone. The radiographic appearance may suggest traumatic new bone formation, a stress fracture, or even an osteosarcoma. However, the dense cortical thickening has a characteristic “dripping candle wax” appearance (irregular cortical hyperostosis), and image stability on serial x-rays is sufficient to make the diagnosis without biopsy.
Discomfort is treated with analgesics or anti-inflammatory agents, and serial x-rays are done over time. Attempts at surgical removal or en bloc resection for severely painful lesions have usually not been successful, and on very rare occasions, amputation is needed.
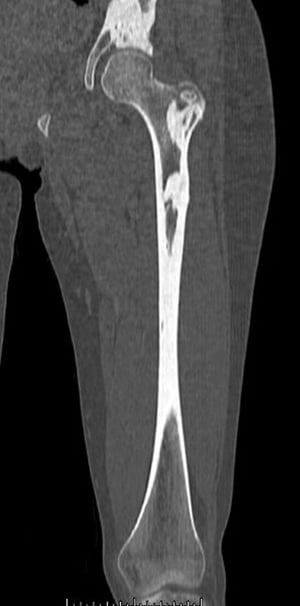
Image courtesy of Michael J. Joyce, MD, and David M. Joyce, MD.
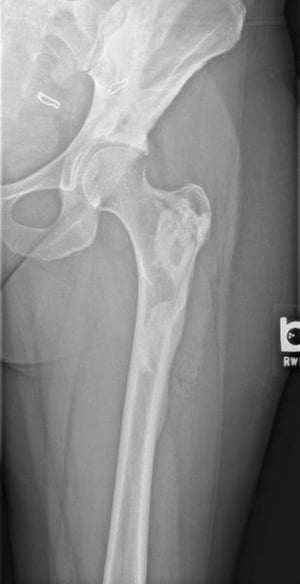
Image courtesy of Michael J. Joyce, MD, and David M. Joyce, MD.
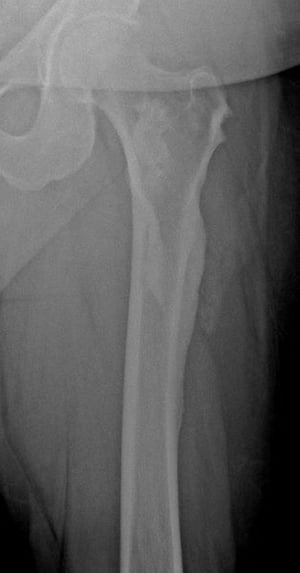
Image courtesy of Michael J. Joyce, MD, and David M. Joyce, MD.

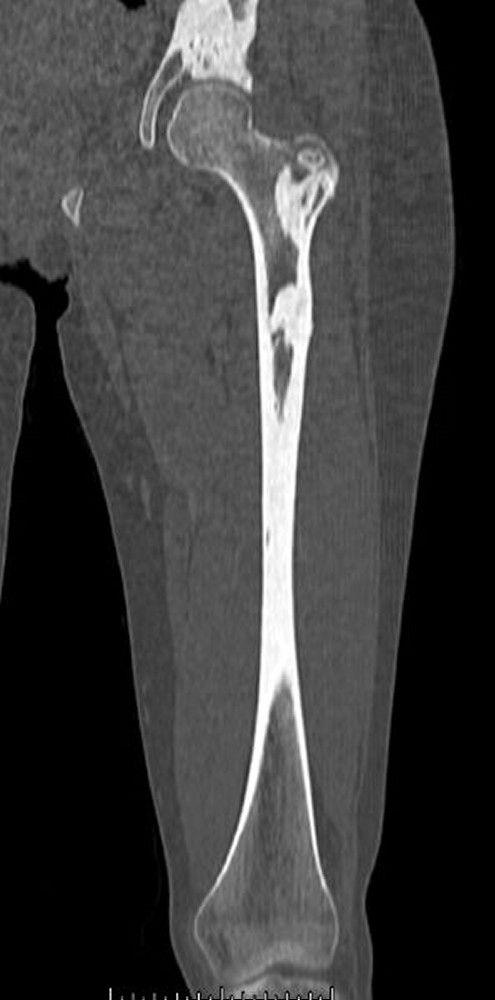
Image courtesy of Michael J. Joyce, MD, and David M. Joyce, MD.

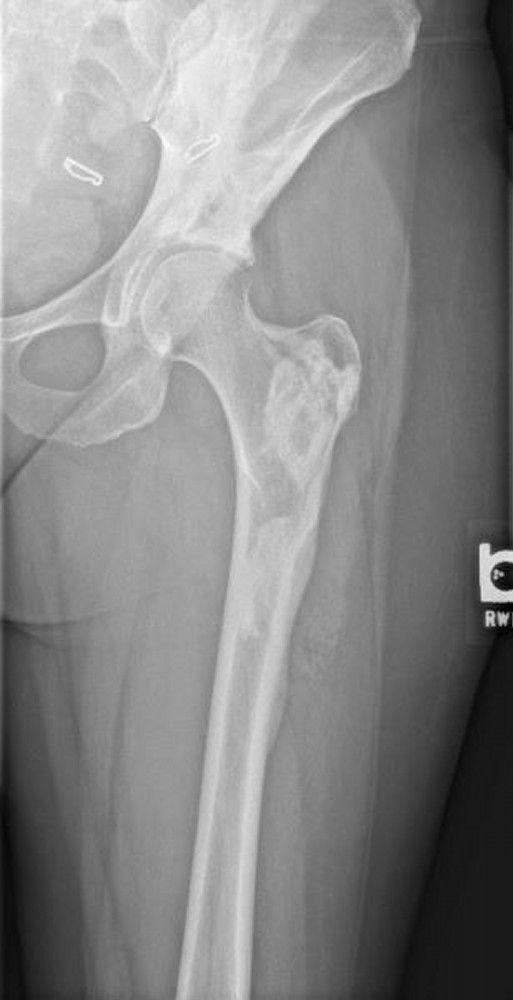
Image courtesy of Michael J. Joyce, MD, and David M. Joyce, MD.

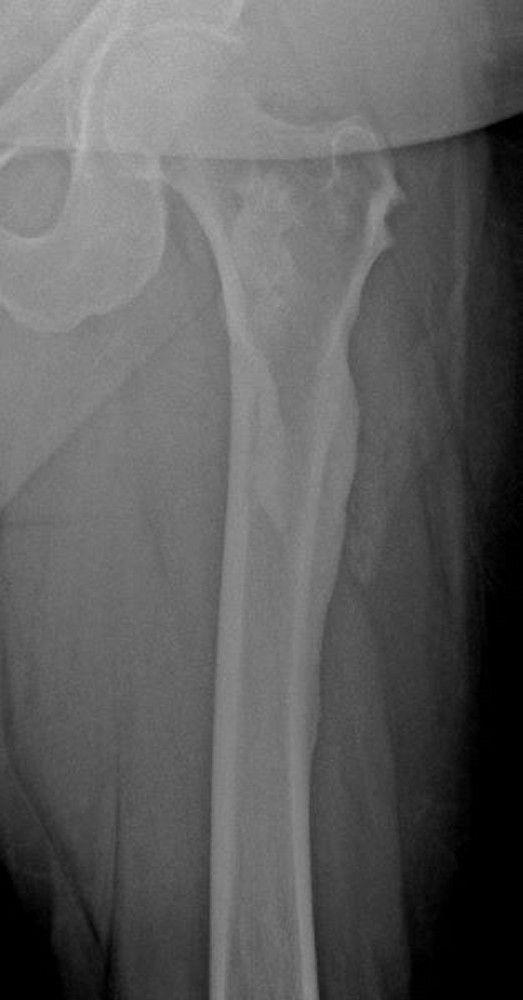
Image courtesy of Michael J. Joyce, MD, and David M. Joyce, MD.
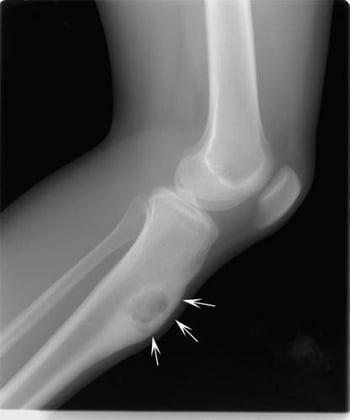
Nonossifying fibroma (fibrous cortical defect, fibroxanthoma)
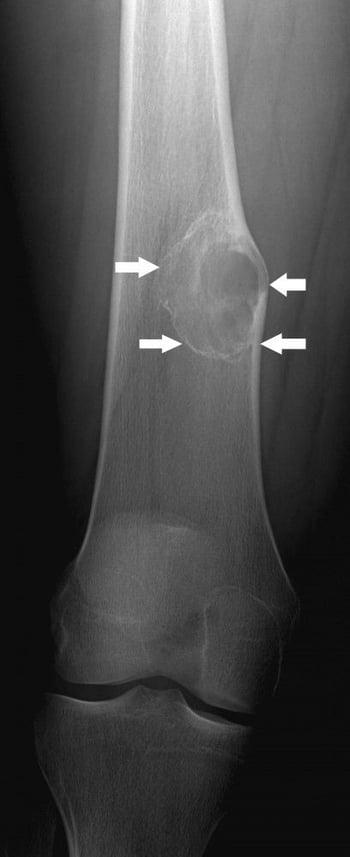
Nonossifying fibroma is a benign fibrous lesion of bone that appears as a well-defined lucent cortical lesion on x-ray. A very small nonossifying fibroma is called a fibrous cortical defect. These lesions are developmental defects in which parts of bone that normally ossify are instead filled with fibrous tissue. They commonly affect the metaphyses, and the most commonly affected sites are, in order, the distal femur, distal tibia, and proximal tibia. They can progressively enlarge and become multiloculated. Nonossifying fibromas are common among children. Most lesions eventually ossify and undergo remodeling, often resulting in dense, sclerotic areas. However, some lesions enlarge.
Small nonossifying fibromas are asymptomatic. However, lesions that involve nearly 50% of the bone diameter tend to cause pain and increase the risk of pathologic fracture.
Nonossifying fibromas are generally first noted incidentally on imaging studies (eg, after trauma). They typically are radiolucent, single, < 2 cm in diameter, and have an oblong lucent appearance with a well-defined sclerotic border in the cortex. They can also be multiloculated.
Small nonossifying fibromas require no treatment and limited follow-up. Lesions that cause pain or are close to 50% of the bone diameter may warrant curettage and bone grafting to decrease risk of a pathologic fracture through the lesion.

Osteoid osteoma
Characteristic appearance on imaging studies is a small radiolucent zone surrounded by a larger sclerotic zone. If a tumor is suspected, a total body technetium-99m bone scan should be done; an osteoid osteoma appears as an area of increased uptake, although many other bone lesions do so as well. CT with fine image sequences or MRI is also done and is most helpful in distinguishing the lesion. Plain films or fine cut CT scan may show a classic "bull's eye" appearance of the true nidus of the tumor surrounded by reactive bone.
Ablation of the small radiolucent zone with percutaneous radiofrequency energy provides permanent relief in most cases. Most osteoid osteomas are treated by an interventional musculoskeletal radiologist using percutaneous techniques and anesthesia. Less often, osteoid osteomas are surgically curetted or excised. Surgical removal may be preferred when the osteoid osteoma is near a nerve or close to the skin (eg, spine, hands, feet) because the heat produced by radiofrequency ablation may cause damage.
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
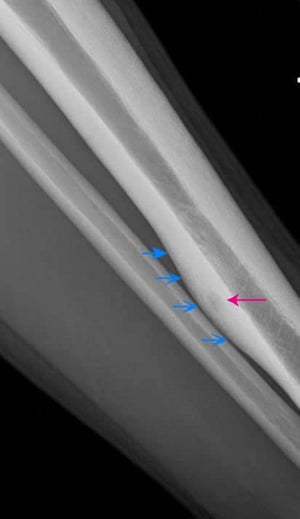
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.

Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.

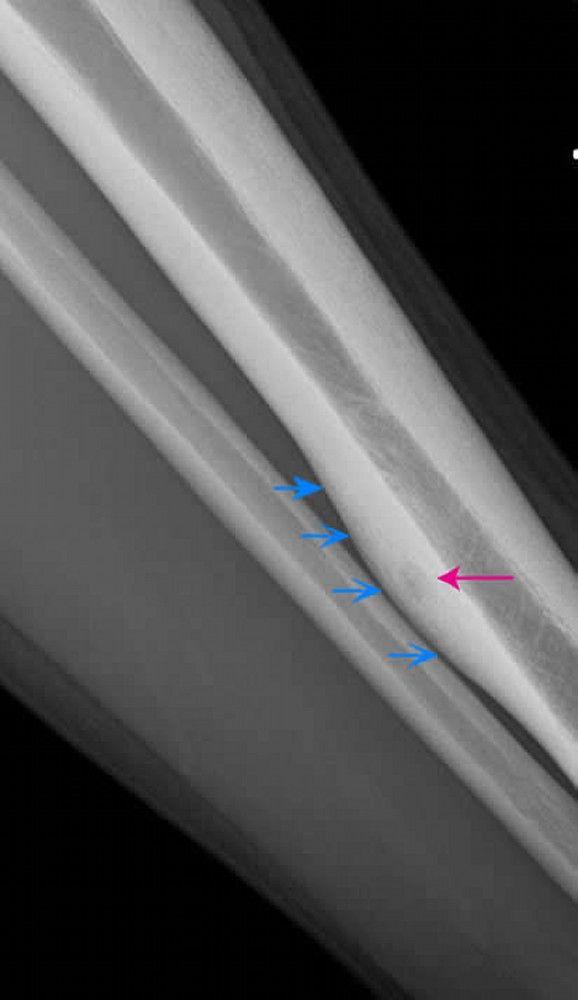
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.

Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
Osteoblastoma
Osteoblastoma is a rare benign tumor that consists of tissue histologically similar to that of an osteoid osteoma. Some experts simply consider them large osteoid osteomas (> 2 cm). Osteoblastoma is much more common among males and appears typically between ages 10 and 35. The tumor develops in the bone of the spine, legs, hands, and feet. It is a slow-growing tumor that destroys normal bone. This tumor is painful.
Imaging is with plain films, CT, and MRI. Usually an open biopsy is warranted to make an accurate diagnosis of osteoblastoma.
Treatment of osteoblastoma requires surgery, often curettage and bone grafting. The local recurrence rate for lesions that are treated with intralesional curettage may be as high as 10 to 20%. More aggressive-appearing lesions are treated with surgical en bloc resection and bony reconstruction. A variation called aggressive osteoblastoma is similar to osteosarcoma both radiographically and histologically.
Osteochondroma
Osteochondromas (osteocartilaginous exostoses), the most common benign bone tumor, may arise from any bone but tend to occur near the ends of long bones. These tumors manifest most often in people aged 10 to 20 and may be single or multiple. Multiple osteochondromas tend to run in families. Secondary malignant chondrosarcoma develops in well under 1% of patients with single osteochondromas, but in about 10% of patients with multiple osteochondromas. Patients with multiple hereditary osteochondromas (formerly called multiple hereditary exostoses- type 1 and type 2) have more tumors and are more likely to develop a chondrosarcoma than patients with a single osteochondroma. Osteochondromas rarely cause the bone to fracture.
On imaging studies, the lesion appears as a bony prominence with a cartilage cap (usually < 2 cm) off the surface of the bone with no underlying cortex under the prominence. MRI may be helpful in differentiating among a thick cartilage cap, bursa, or surrounding soft tissue mass. The medullary canal is in continuity with the base of the exostosis. Occasionally, a painful bursa may form over the cartilage cap.
Excision is needed if the tumor is compressing a large nerve or vessel; causes pain (especially when impinging on muscle and creating an inflammatory bursa); disturbs growth; or on imaging study has a destructive appearance, soft tissue mass, or thickened cartilaginous cap (> 2 cm) suggesting transformation into malignant chondrosarcoma. An enlarging tumor in an adult should raise concern of chondrosarcoma and the possible need for excision or biopsy.

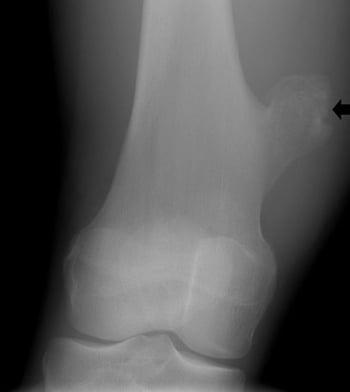
Osteoma
Osteomas are areas of uniformly dense bone that appear benign. They are most common in the skull and area around the paranasal bones and rarely occur in the axial skeleton or pelvis. Small osteomas are called bone islands. These are not painful and often noted incidentally as bone densities with smooth margins on x-rays. Diagnosis is based on radiographic appearance. On nuclear bone scan, the radiographic dense area may show little or no increased uptake. If the diagnosis is questionable, MRI imaging can help. Osteomas do not require biopsy and can be followed periodically with plain films.
Osteopoikilosis
Osteopoikilosis is a benign sclerosing dysplastic bone disease with autosomal dominant inheritance characterized by numerous asymptomatic dense bone islands throughout the skeleton. It usually involves the axial skeleton (skull, spine, pelvis) and ribs but sometimes involves the extremities, particularly the proximal femurs. Osteopoikilosis causes no symptoms.
Osteopoikilosis is usually noted as an incidental finding on x-ray. Diagnosis of osteopoikilosis is based on radiographic appearance, and patients often have earlier films demonstrating that these dense multiple bone islands have retained their same typical appearance over time. These bone islands show little or no activity on nuclear bone scan. Serial films can be done to confirm no change over time. Rarely patients with osteopoikilosis have melorheostosis. In middle age and older adults, small osteoblastic lesions of metastatic disease from carcinomas (breast, prostate, lung) must be considered in the differential diagnosis, but metastatic lesions are usually larger and have more irregular margins, while osteopoikilosis lesions are more numerous in periarticular areas.

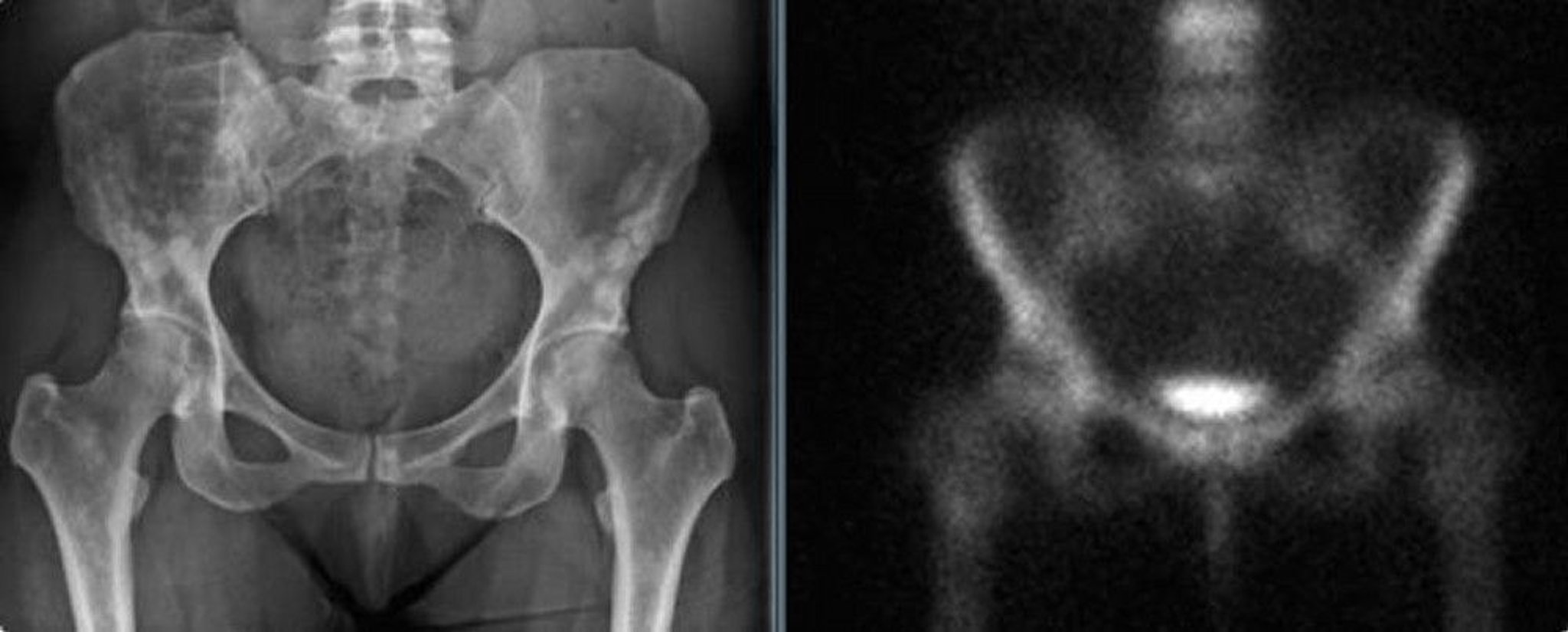
Images courtesy of Michael J. Joyce, MD, and David M. Joyce, MD.
Unicameral bone cyst
Simple unicameral bone cysts occur in the long bones starting distal to the epiphyseal plate in children. The cyst is fluid-filled. It causes the cortex to thin and predisposes the area to a buckle-like pathologic fracture, which is usually how the cyst is recognized.
Plain x-rays are usually diagnostic. Simple unicameral bone cysts typically appear as well-marginated lesions without reactive sclerosis or an expansile cortex. If the cyst has a minor fracture, a bone fragment from the thin shell may fall to the bottom of the fluid-filled cyst. The result is the classic "fallen leaf appearance" on x-ray.
Smaller cysts sometimes heal without treatment. A nondisplaced fracture through small cysts may be a stimulus for healing. However, if the cyst is > 85% of the diameter of the bone or the bony shell is < 0.5 mm, there is a higher risk of pathological fracture. Larger cysts, particularly in children, may require curettage and bone grafting; however, many respond to injections of corticosteroids, demineralized bone matrix, or synthetic bone substitutes. The response may be variable and may require multiple injections. Regardless of treatment, cysts persist in about 10 to 15% of patients.