


Behçet disease is a multisystem, relapsing, chronic vasculitic disorder with mucosal inflammation. Common manifestations include recurrent oral ulcers, ocular inflammation, genital ulcers, and skin lesions. The most serious manifestations are blindness, neurologic or gastrointestinal manifestations, venous thromboses, and arterial aneurysms. Diagnosis is clinical, using international criteria. Treatment is mainly symptomatic but may involve corticosteroids with or without other immunosuppressants for more severe manifestations.
(See also Overview of Vasculitis.)
Behçet disease is an inflammatory disorder that can include a vasculitis of small and large arteries and/or veins. Arterial and venous thrombosis may occur as well.
The disease occurs nearly equally in men and women but tends to be more severe in men, typically beginning during their 20s. Occasionally, the disease develops in children. Incidence varies by location. Behçet disease is most common along the silk route from the Mediterranean to China; it is uncommon in the US.
The cause of Behçet disease is unknown. Immunologic (including autoimmune) and viral or bacterial triggers have been suggested, and HLA-B51 is a major risk factor. Prevalence of an HLA-B51 allele is > 15% among people from Europe, the Middle East, and the Far East but is low or absent among people from Africa, Oceania, and South America.
Neutrophil infiltration is detected in biopsy specimens from oral aphthous ulcers and erythema nodosum and pathergy lesions, but no histologic changes are pathognomonic.
Symptoms and Signs of Behçet Disease
Mucocutaneous
Almost all patients have recurrent, painful oral ulcers resembling those of aphthous stomatitis; in most, these ulcers are the first manifestations. The ulcers are round or oval, 2 to 10 mm in diameter, and shallow or deep with a central yellowish necrotic center; they can occur anywhere in the oral cavity, often in clusters. Ulcers last 1 to 2 weeks. Similar ulcers occur on the penis and scrotum, on the vulva where they are painful, or in the vagina where they may cause little or no pain.
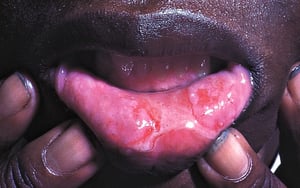
© Springer Science+Business Media
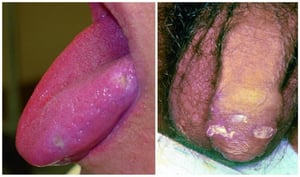
© Springer Science+Business Media
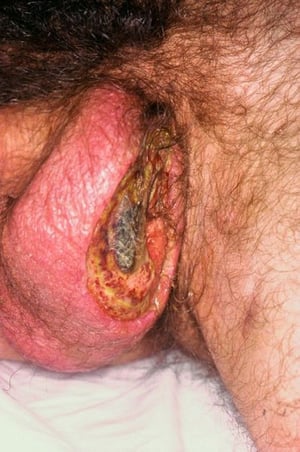
Image courtesy of Karen McKoy, MD.

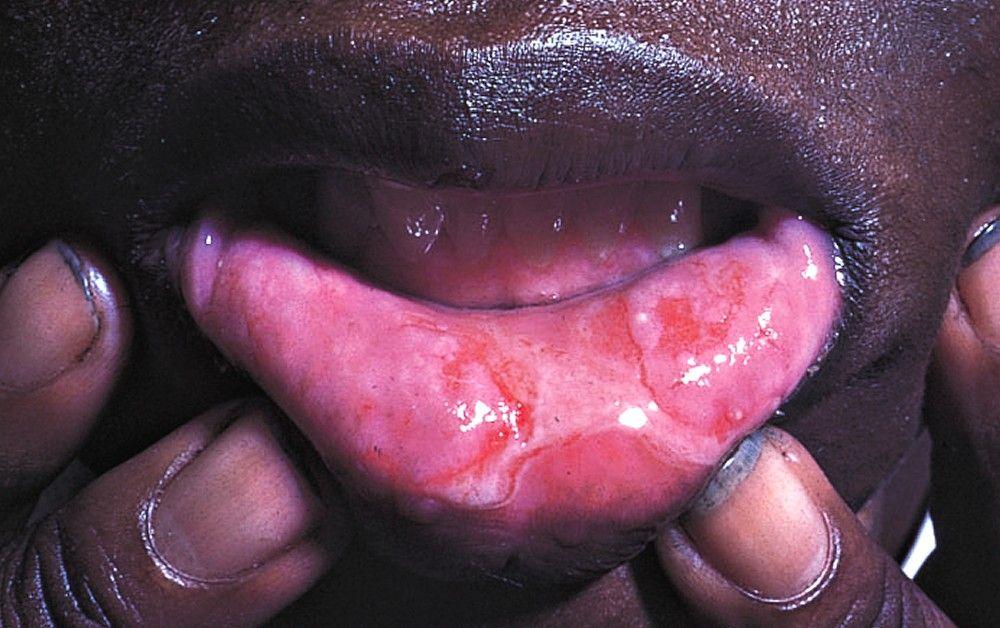
© Springer Science+Business Media

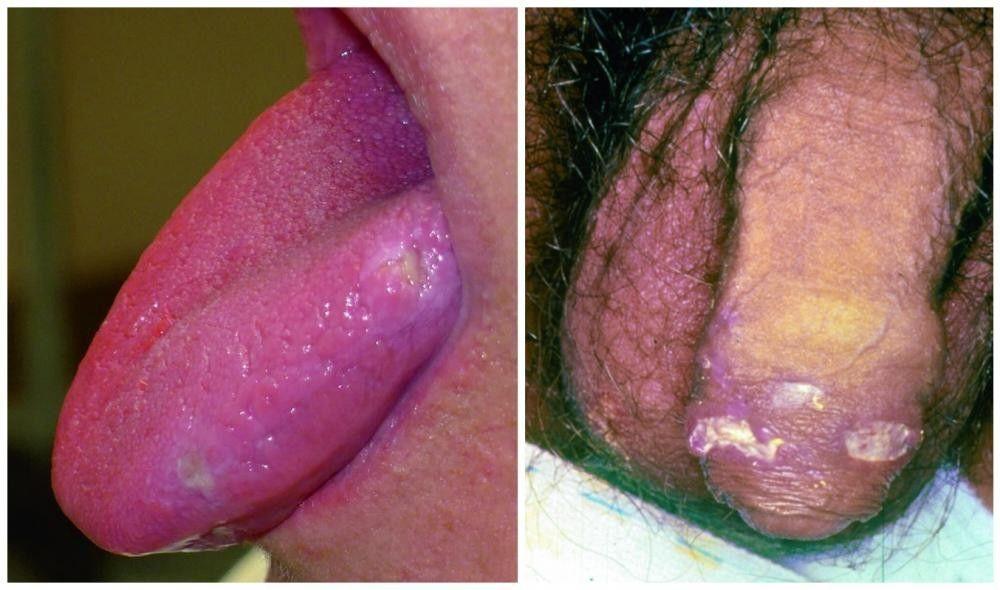
© Springer Science+Business Media

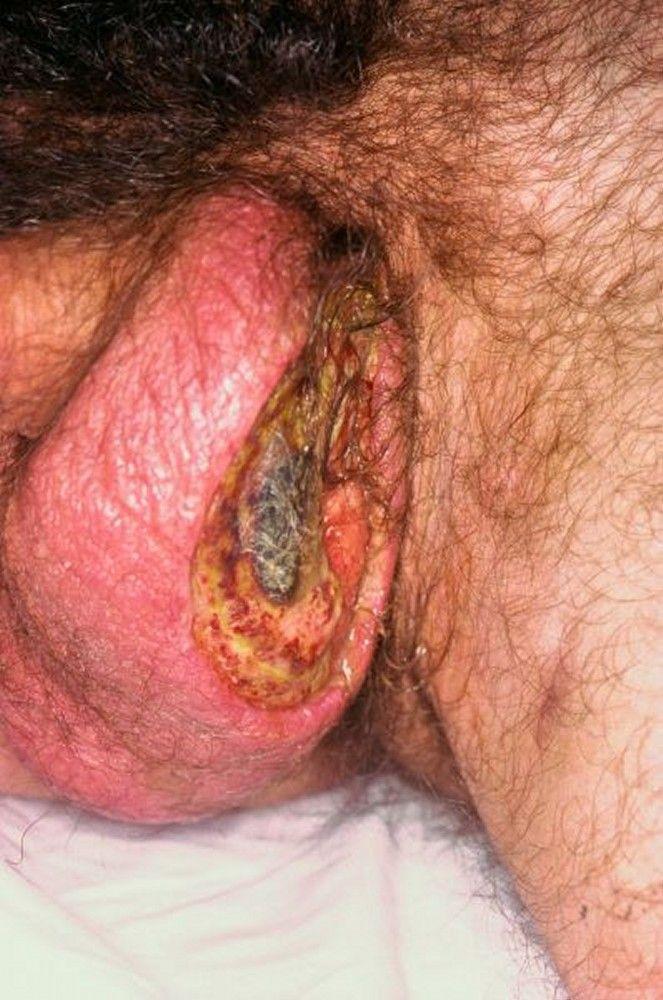
Image courtesy of Karen McKoy, MD.
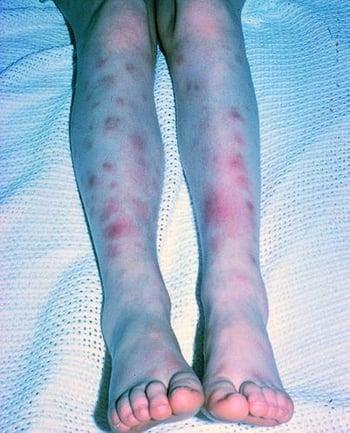
Cutaneous lesions are common and may include acneiform lesions, nodules, erythema nodosum, superficial thrombophlebitis, pyoderma gangrenosum–type lesions, and palpable purpura.

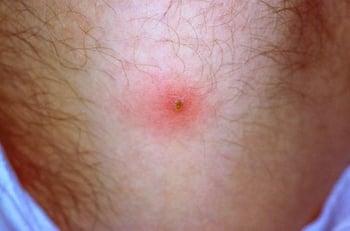
Pathergy (an erythematous papular or pustular response to local skin injury) is defined as a papule > 2 mm that appears 24 to 48 hours after oblique insertion of a 20- to 25-gauge needle into the skin.

Ocular
The eyes are affected in 25 to 75% of patients. Eye manifestations may be associated with neurologic manifestations. The following may occur:
Relapsing uveitis or iridocyclitis (most common) often manifests as pain, photophobia, and red eye.
Hypopyon (a layer of pus visible in the anterior chamber) may occur.
Uveitis is typically bilateral and episodic, often involves the entire uveal tract (panuveitis), and may not resolve completely between episodes.
Choroiditis, retinal vasculitis, vascular occlusion, and optic neuritis may irreversibly impair vision and even progress to blindness.

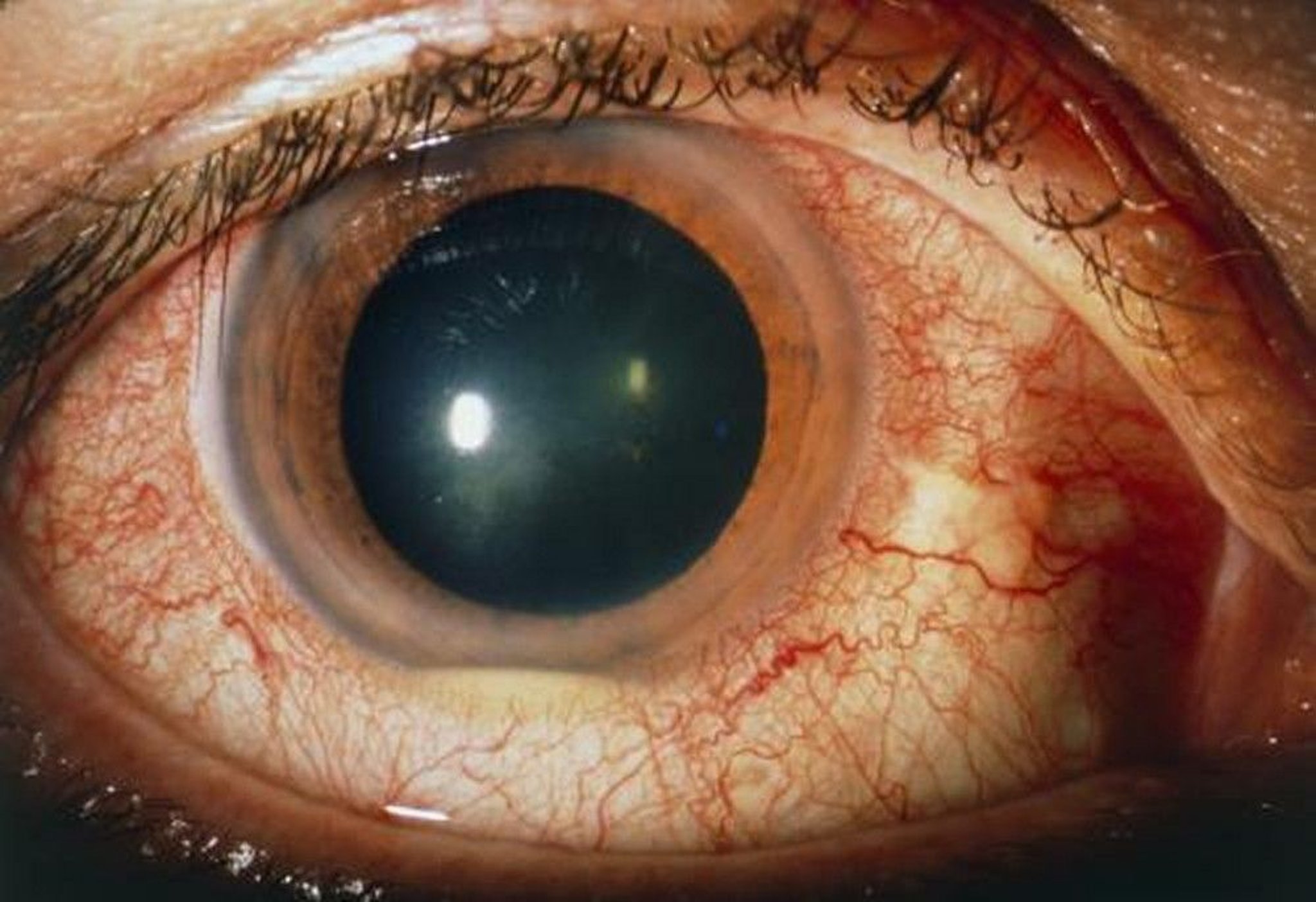
SUE FORD/SCIENCE PHOTO LIBRARY

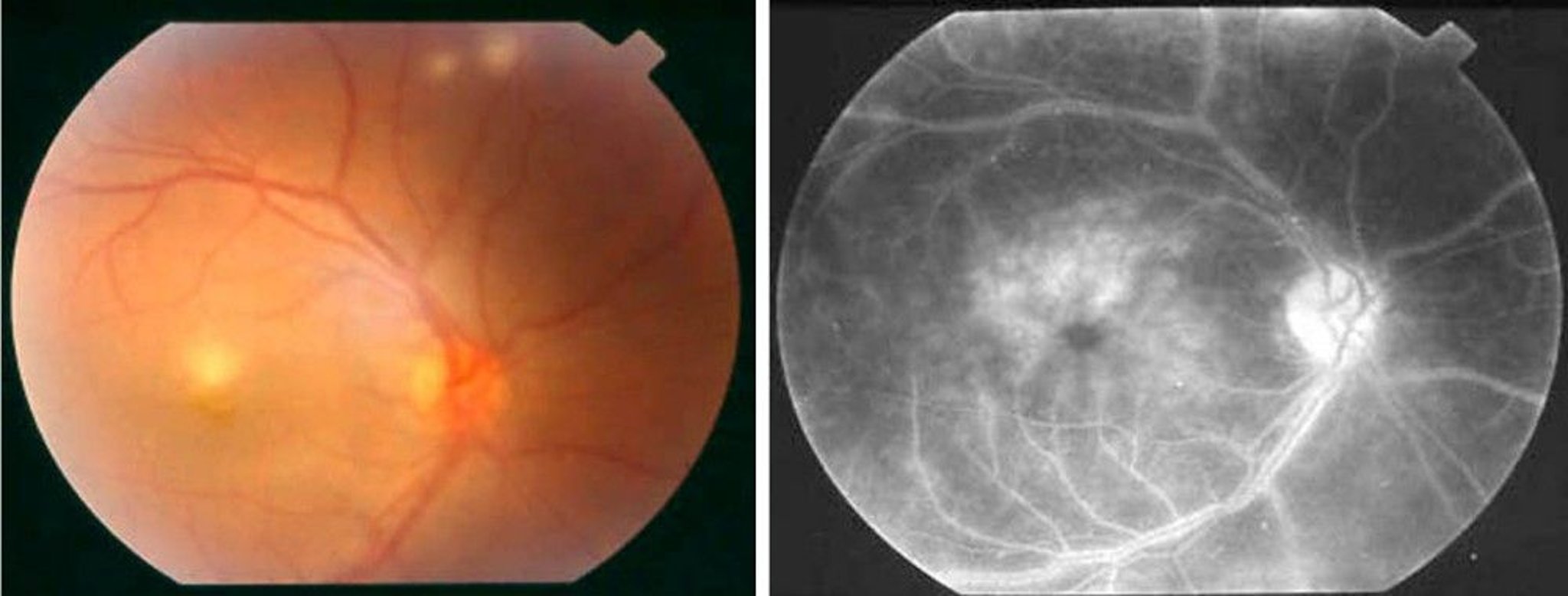
© Springer Science+Business Media
Musculoskeletal
Relatively mild, self-limiting, and nondestructive arthralgias or frank arthritis, especially in the knees and other large joints, occur in 50% of patients. Sacroiliac inflammation can occur.
Vascular
Perivascular and endovascular inflammation may develop in arteries and veins. In arteries, thrombosis, aneurysm, pseudoaneurysm, hemorrhage, and stenosis can develop. Large-vessel arterial involvement is recognized during life in 3 to 5% of patients; however, at autopsy, one third of patients have evidence of large-vessel involvement that was asymptomatic during life. Aortic and pulmonary artery aneurysms can rupture. In situ thrombosis can cause pulmonary artery occlusion. Hemoptysis may occur if fistulas develop between the pulmonary artery and bronchus.
Venous involvement can cause superficial and deep venous thromboses. More than one vein may be affected, including the inferior and superior vena cava, the hepatic veins (causing Budd-Chiari syndrome), and the dural venous sinuses.
In situ arterial or venous thromboses, aneurysms, and pseudoaneurysms are more common than stenoses and occlusions.
Neurologic and psychiatric
Central nervous system involvement is less common but is serious. Onset may be sudden or gradual. The first manifestations may be parenchymal involvement with pyramidal signs, small-vessel disease with a multiple sclerosis–like pattern, nonparenchymal involvement with aseptic meningitis or meningoencephalitis, or dural sinus thrombosis. Aseptic meningitis, in the characteristic clinical setting, can suggest the diagnosis.
Psychiatric disorders including personality changes and dementia may develop years later. Peripheral neuropathy, common in other vasculitic disorders, is uncommon in Behçet disease.
Gastrointestinal (GI)
Abdominal discomfort, abdominal pain, and diarrhea with intestinal ulcers, occurring primarily in the ileum and colon and closely resembling Crohn disease, may occur.
Constitutional
Fever and malaise may occur.
Diagnosis of Behçet Disease
Clinical criteria
Behçet disease should be suspected in young adults with recurrent oral aphthous ulcers, unexplained ocular findings, or genital ulcers. Diagnosis of Behçet disease is clinical and often delayed because many of the manifestations are nonspecific and can be insidious.
International criteria for diagnosis include recurrent oral ulcers (3 times in 1 year) and 2 of the following:
Recurrent genital ulcers
Eye lesions
Skin lesions
Positive pathergy test with no other clinical explanation
A positive pathergy test consists of the appearance of an erythematous induration with a sterile pustule in the skin 24 to 48 hours after the insertion of a sterile needle into the skin of the forearm.
Differential diagnosis
Differential diagnosis includes
Behçet disease has no single pathognomonic finding but may be distinguished by its combinations of relapsing symptoms with spontaneous remissions and multiple organ involvement, particularly in patients with recurrent, deep mucosal ulcers.
Prognosis for Behçet Disease
Behçet disease typically has a waxing and waning course characterized by exacerbations and remissions. Prognosis tends to be worse in young men. Risk also appears to be higher if patients have an HLA-B51 allele. Mucocutaneous and ocular lesions and arthralgias are often worse early in the disease. Central nervous system and large-vessel manifestations, if they develop, typically occur later. Occasionally, the disease results in death, usually due to neurologic, vascular (eg, aneurysms), or gastrointestinal manifestations. Risk of death is highest for young men and patients with arterial disease or a high number of flare-ups. Many patients eventually go into remission.
Treatment of Behçet Disease
Treatment of Behçet disease depends on the clinical manifestations. Treatment recommendations are limited by incomplete data from clinical studies (eg, cross-sectional studies, usually not prospective, limited statistical power).
Immunosuppressants, including anti-TNF drugs, improve the prognosis for patients with vascular involvement. Immunosuppressants help prevent recurrence of venous thrombosis, but it is unclear whether anticoagulation does. Anticoagulation is contraindicated in patients with pulmonary arterial aneurysms.
Mucosal disease
Colchicine, which was hypothesized to decrease the need for later immunosuppressive therapy when used early in the disease course, has not been shown to do so.
glucose-6-phosphate dehydrogenase (G6PD) deficiency.
colchicine
Ocular disease
Azathioprine is also useful for mucocutaneous lesions and arthralgia.
azathioprine to treat refractory uveitis.
Refractory or life-threatening disease
cyclophosphamide than with azathioprine.
Key Points
Behçet disease is a relapsing inflammatory disorder characterized by prominent mucosal inflammation often with vasculitis of large and small vessels.
Among the many organ systems involved, oral and genital ulcers, skin lesions, aseptic meningitis, and ocular findings, particularly in combination, are very characteristic.
Diagnose based on specific clinical criteria.
Risk factors for early death are male sex, frequent disease flare-ups, and arterial complications (eg, thrombosis, aneurysms, pseudoaneurysms).






