


The duodenum can be obstructed by atresia, stenosis, and pressure due to an extrinsic mass.
(See also Overview of Congenital Gastrointestinal Anomalies.)
Duodenal atresia
Duodenal atresia is the 2nd most common atresia of the gastrointestinal (GI) tract. The estimated incidence is 1 in 10,000 live births (1). Duodenal atresia is due to the failure of canalization of the embryonic duodenum. This failure may be related to an ischemic event or genetic factors.
Duodenal atresia, unlike other intestinal atresias, is commonly associated with other congenital anomalies; approximately 25 to 40% of infants with duodenal atresia have Down syndrome (1). Other associated anomalies include VACTERL (vertebral anomalies, anal atresia, cardiac malformations, tracheoesophageal fistula, esophageal atresia, renal anomalies and radial aplasia, and limb anomalies), malrotation, annular pancreas, biliary tract abnormalities, and mandibulofacial anomalies.
Diagnosis of duodenal atresia is suspected prenatally if there is polyhydramnios and/or a dilated stomach. Prenatal ultrasonography can detect a double bubble sign (a large gastric bubble and a smaller proximal duodenal bubble) in up to 80% of cases (2).

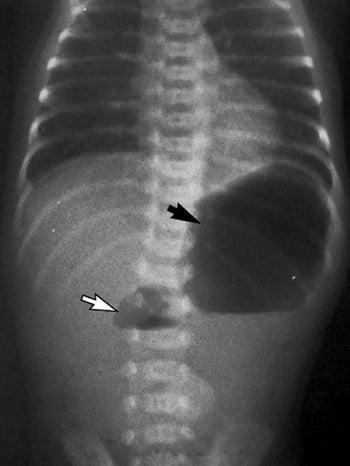
Postnatally, infants with duodenal atresia present with feeding difficulties and emesis that may be bilious. The diagnosis is suspected by symptoms and classic double-bubble x-ray findings—one bubble is in the stomach and the other is in the proximal duodenum; little to no air is in the distal gut. Although an upper GI series provides definitive diagnosis, it must be done carefully by a radiologist experienced with doing this procedure on children to avoid aspiration and is not typically necessary if surgery is to be done immediately. If surgery is to be delayed (eg, because of other medical issues, such as respiratory distress syndrome or the need to stabilize the infant), a contrast enema should be done to confirm that the double-bubble sign is not due to malrotation.
Once the disorder is suspected, infants should receive nothing by mouth, and a nasogastric tube should be placed to decompress the stomach.
Surgery is the definitive therapy.
Duodenal stenosis
This anomaly occurs less commonly than duodenal atresia but manifests in a similar fashion and requires surgery. It too is frequently associated with Down syndrome.
Choledochal cyst
A choledochal cyst may obstruct the duodenum by extrinsic pressure. The incidence ranges from approximately 1 in 100,000 people in the United States or Europe to 1 in 13,000 in Japan (3). Over half of reported cases occur in Japan. There is some evidence that the incidence of choledochal cysts is increasing.
Infants with choledochal cyst classically present with a triad of abdominal pain (a very difficult finding to infer in the neonate), right upper quadrant mass, and jaundice. If the cyst is large, it may also manifest with variable degrees of duodenal obstruction. Neonates can present with cholestasis. In some cases, pancreatitis is an associated finding.
Choledochal cyst is most commonly diagnosed by ultrasonography. These cysts can be further defined by using magnetic resonance cholangiopancreatography, endoscopic retrograde cholangiopancreatography (ERCP), or endoscopic ultrasonography.
Treatment of choledochal cyst is surgical and requires complete excision of the cyst because of the high risk (20 to 30%) of developing cancer in the cyst remnants (4). The surgical procedure most commonly used is a Roux-en-Y hepaticojejunostomy. This procedure is typically done laparoscopically. Some recent studies have noted that the outcome may be enhanced with robotic-assisted surgery.
Annular pancreas
Annular pancreas is a rare congenital anomaly (5 to 15 per 100,000 live births [5]), often associated with Down syndrome, in which pancreatic tissue encircles the 2nd portion of the duodenum, causing a duodenal obstruction.
About two thirds of affected people remain asymptomatic. Of those who develop symptoms, most present in the neonatal period, but manifestation may be delayed until adulthood. Neonates present with feeding problems and emesis that may be bilious.
The diagnosis of annular pancreas can be suggested by an x-ray of the abdomen showing the same double-bubble sign seen in duodenal atresia. The diagnosis can also be made by an upper GI series and is more definitively made with CT or magnetic resonance cholangiopancreatography. ERCP can be done in older children.
Treatment of annular pancreas is surgical bypass of the annular pancreas with duodenoduodenostomy, duodenojejunostomy, or gastrojejunostomy. Resection of the pancreas should be avoided because of the potential complications of pancreatitis and pancreatic fistula development.
References
1. Bethell GS, Long AM, Knight M, Hall NJ; BAPS-CASS: Congenital duodenal obstruction in the UK: a population-based study. Arch Dis Child Fetal Neonatal Ed 105(2):178-183, 2020. doi:10.1136/archdischild-2019-317085
2. Engwall-Gill AJ, Zhou AL, Penikis AB, et al: Prenatal sonography in suspected proximal gastrointestinal obstructions: Diagnostic accuracy and neonatal outcomes. J Pediatr Surg 58(6):1090-1094, 2023. doi: 10.1016/j.jpedsurg.2023.02.029. Epub 2023 Feb 17. PMID: 36907770
3. Soares KC, Kim Y, Spolverato G, et al: Presentation and clinical outcomes of choledochal cysts in children and adults: a multi-institutional analysis. JAMA Surg 150(6):577-584, 2015. doi:10.1001/jamasurg.2015.0226
4. Søreide K, Søreide JA: Bile duct cyst as precursor to biliary tract cancer. Ann Surg Oncol 14(3):1200-1211, 2007. doi:10.1245/s10434-006-9294-3
5. Alkhayyat M, Bachour S, Abou Saleh M, et al: The epidemiology of annular pancreas in the United States: a population-based study. J Clin Gastroenterol 56(2):186-191, 2022. doi:10.1097/MCG.0000000000001531






