


Idiopathic pulmonary fibrosis (IPF), the most common form of idiopathic interstitial pneumonia, causes progressive pulmonary fibrosis. Symptoms and signs develop over months to years and include exertional dyspnea, cough, and fine (Velcro) crackles. Diagnosis is based on history, physical examination, high-resolution CT, and/or lung biopsy, if necessary. Treatment may include antifibrotic drugs and oxygen therapy. Most patients deteriorate; median survival is about 3 years from diagnosis.
Idiopathic pulmonary fibrosis, identified histologically as usual interstitial pneumonia, accounts for most cases of idiopathic interstitial pneumonia. IPF affects men and women > 50 in a ratio of 2:1, with a markedly increased incidence with each decade of age. Current or former cigarette smoking is most strongly associated with the disorder. There is some genetic predisposition; familial clustering occurs in up to 20% of cases.
Etiology of Idiopathic Pulmonary Fibrosis
A combination of environmental, genetic, and other unknown factors probably contribute to alveolar epithelial cell dysfunction or reprogramming, which leads to abnormal fibroproliferation in the lung. There is ongoing research into the contributions of genetics, environmental stimuli, inflammatory cells, the alveolar epithelium, mesenchyme, and matrix.
Pathology of Idiopathic Pulmonary Fibrosis
The key histologic findings of idiopathic pulmonary fibrosis are subpleural fibrosis with sites of fibroblast proliferation (fibroblast foci) and dense scarring, alternating with areas of normal lung tissue (heterogeneity). Scattered interstitial inflammation occurs with lymphocyte, plasma cell, and histiocyte infiltration. Cystic abnormality (honeycombing) occurs in all patients and increases with advanced disease. A similar histologic pattern uncommonly occurs in cases of interstitial lung diseases of known etiology (see table Key Features of Idiopathic Interstitial Pneumonias).
Symptoms and Signs of Idiopathic Pulmonary Fibrosis
Symptoms and signs of idiopathic pulmonary fibrosis typically develop over 6 months to several years and include dyspnea on exertion and nonproductive cough. Constitutional symptoms, such as low-grade fever and myalgias, are uncommon. The classic sign of IPF is fine, dry, inspiratory crackles (Velcro crackles) at both bases. Clubbing is present in about 50% of cases. The remainder of the examination is normal until disease is advanced, at which time signs of pulmonary hypertension and right ventricular systolic dysfunction may develop.
Diagnosis of Idiopathic Pulmonary Fibrosis
High-resolution CT (HRCT)
Sometimes surgical lung biopsy
Diagnosis of idiopathic pulmonary fibrosis is suspected in patients with subacute dyspnea, nonproductive cough, and Velcro crackles on chest examination. However, IPF is commonly overlooked initially because of clinical similarities to other more common diseases, such as bronchitis, asthma, and heart failure.
Diagnosis requires HRCT and in some cases lung biopsy.
Chest x-ray typically shows diffuse reticular opacities in the lower and peripheral lung zones. Small cystic lesions (honeycombing) and dilated airways due to traction bronchiectasis are additional findings.
HRCT shows diffuse, patchy, subpleural, reticular opacities with irregularly thickened interlobular septa and intralobular lines; subpleural honeycombing; and traction bronchiectasis. This is referred to as the usual interstitial pneumonia (UIP) pattern. Ground-glass opacities affecting > 30% of the lung suggest an alternative diagnosis.

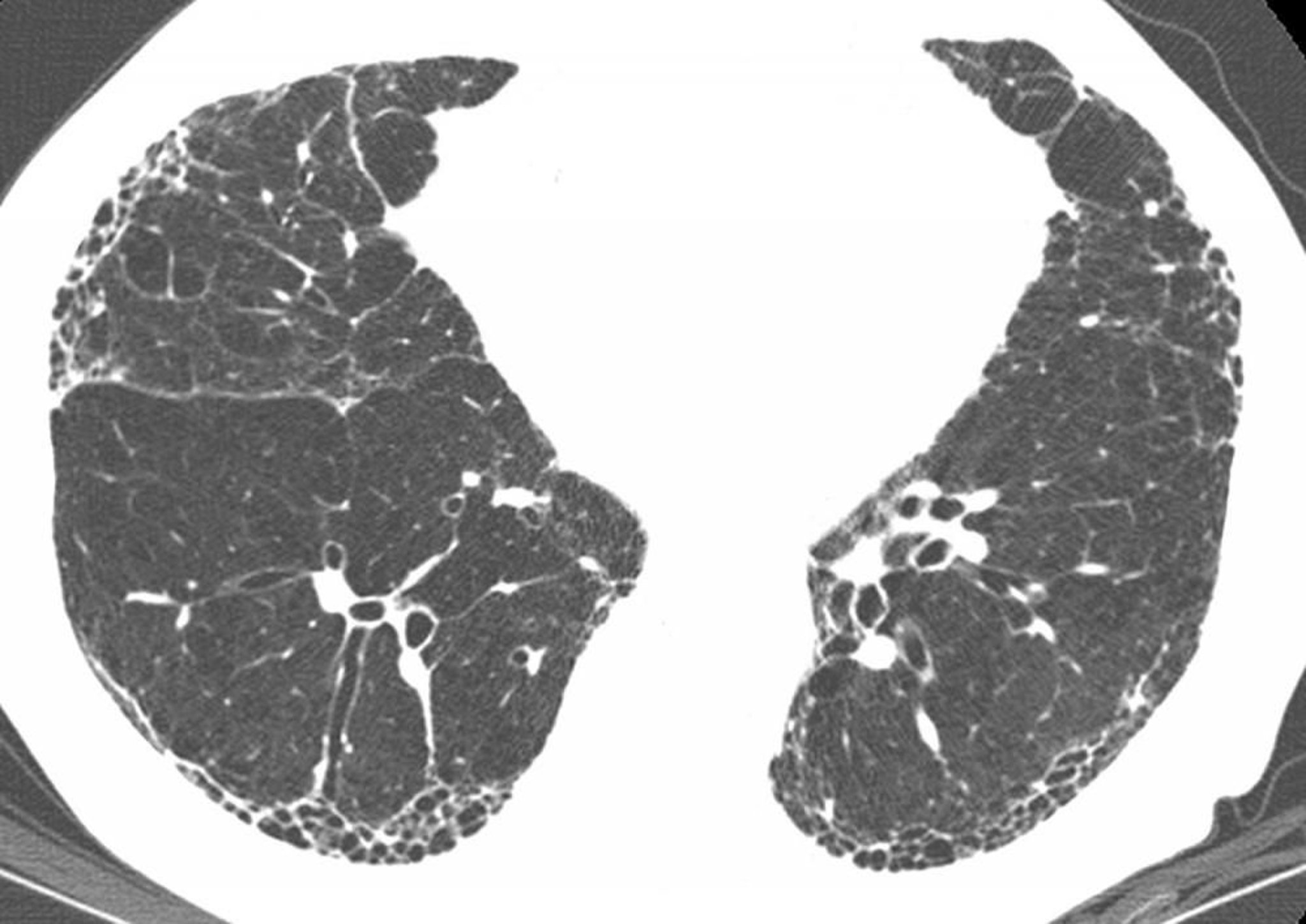
Image courtesy of Harold R. Collard, MD.
Laboratory testing plays little role in diagnosis, except to exclude potential systemic rheumatic disorders.
Treatment of Idiopathic Pulmonary Fibrosis
Oxygen and pulmonary rehabilitation
Sometimes lung transplantation
1, 2, 3). Supportive measures include oxygen and pulmonary rehabilitation. Patients may find that joining a support group helps reduce the stress of the illness.
Many novel therapies for IPF are under development or being tested as treatments for IPF, and patients should be encouraged to participate in clinical trials when appropriate.
Lung transplantation is successful for otherwise healthy patients with IPF, generally those < 65 years old. These patients should be evaluated for lung transplantation at the time of diagnosis.
Treatment references
1.King TE, Bradford WZ, Castro-Bernardini S, et al: A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Eng J Med 370:2083-2092, 2014.
2. Raghu G, Rochwerg B, Zhang Y, et al: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med 192 (2):e3-e19, Jun 15, 2015.
3. Richeldi L, du Bois RM, Raghu G, et al: Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370:2071–2082, 2014.
Prognosis for Idiopathic Pulmonary Fibrosis
Most patients have moderate to advanced clinical disease at the time of diagnosis and deteriorate despite treatment. The mortality rate is estimated to be 50 to 70% at 5 years. Several prognostic models have been proposed. Among the factors that portend a worse prognosis are older age, male sex, lower forced vital capacity, and lower diffusing capacity for carbon monoxide (DLCO).
Causes of acute deterioration include infections, pulmonary embolism, pneumothorax, and heart failure. Also, acute exacerbations without an identifiable cause may occur. All acute exacerbations have a high morbidity and mortality.
Lung cancer occurs more frequently in patients with IPF, but cause of death is usually respiratory failure. Because of the poor prognosis of IPF, early discussions with the patient and family about advance care planning and end-of-life care are important.
Key Points
Idiopathic pulmonary fibrosis accounts for most idiopathic interstitial pneumonia and tends to affect older people.
Symptoms and signs (eg, subacute dyspnea, nonproductive cough, and Velcro crackles) are nonspecific and usually caused by other, more common disorders.
High-resolution CT can help in diagnosis by showing findings such as diffuse, patchy, subpleural, reticular opacities with irregularly thickened interlobular septa and intralobular lines; subpleural honeycombing; and traction bronchiectasis.
Encourage participation in clinical trials and, if patients are < 65 years and otherwise healthy, consider lung transplantation at the time of diagnosis.
More Information
The following English-language resource may be useful. Please note that THE MANUAL is not responsible for the content of this resource.
Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med 2022;205(9):e18-e47. doi:10.1164/rccm.202202-0399ST






