


Sarcoidosis is an inflammatory disorder resulting in noncaseating granulomas in one or more organs and tissues; etiology is unknown. The lungs and lymphatic system are most often affected, but sarcoidosis may affect any organ. Pulmonary symptoms range from none to cough, exertional dyspnea and, rarely, lung or other organ failure. Diagnosis usually is first suspected because of pulmonary involvement and is confirmed by chest x-ray, biopsy, and exclusion of other causes of granulomatous inflammation. Treatment usually is indicated in symptomatic patients. First-line treatment is corticosteroids. Prognosis is excellent for limited disease but poor for more advanced disease.
Sarcoidosis most commonly affects people aged 20 to 40 years but occasionally affects children and older adults. Worldwide, prevalence is greatest in Black American people and ethnic northern European people, especially Scandinavian people. Disease presentation varies widely by racial and ethnic background, with Black American patients having more frequent extrathoracic manifestations. Sarcoidosis is slightly more prevalent in women.
Löfgren syndrome
Löfgren syndrome manifests as a triad of acute migratory polyarthritis, erythema nodosum, and hilar adenopathy. Fever, malaise, uveitis, and parotitis may also be present. Löfgren syndrome is more common among people of European ancestry. Löfgren syndrome is self-limited. Patients can usually be treated with nonsteroidal anti-inflammatory drugs (NSAIDs) alone. Rate of relapse is low.
Heerfordt syndrome
Heerfordt syndrome (uveoparotid fever) manifests as swelling of the parotid gland (due to sarcoid infiltration), uveitis, chronic fever, and less often palsy of the facial nerve. Heerfordt syndrome can be self-limited. Treatment is the same as for sarcoidosis.
Blau syndrome
Blau syndrome is a sarcoidosis-like disease inherited in an autosomal dominant fashion that manifests in children. It is not known whether Blau syndrome arises through the same mechanism as sarcoidosis diagnosed in adults. In Blau syndrome, children present before the age of 4 years with arthritis, rash, and uveitis. Blau syndrome is often self-limited. Symptoms usually are relieved with NSAIDs.
Etiology of Sarcoidosis
Sarcoidosis is thought to be due to an exaggerated inflammatory response to an environmental antigen in a genetically susceptible person. Proposed triggers include
Propionibacterium acnes and mycobacteria (potentially the Mycobacterium tuberculosis catalase-peroxidase [mKatG] protein)
Mold or mildew and certain unidentified substances present in workplaces with musty odors
Pesticides, particularly those containing aluminum compounds
Tobacco use is inversely correlated with sarcoidosis.
Evidence supporting genetic susceptibility includes the following:
Higher rate of disease concordance in monozygotic than dizygotic twins
Increased prevalence of sarcoidosis (about 3.6 to 9.6%) among 1st- or 2nd-degree relatives of patients who have sarcoidosis
Fivefold increase in relative risk of developing sarcoidosis in siblings of patients who have sarcoidosis
Identification of several possible human leukocyte antigen (HLA) and non-HLA genes associated with sarcoidosis risk, course, and phenotypes
For example the HLA-DRB1*03/DQB1*02 haplotype is associated with Löfgren syndrome and predicts excellent prognosis, in contrast to HLA-DRB1*15/HLA DQB1*0602, which predicts persistent disease.
Pathophysiology of Sarcoidosis
The unknown antigen triggers a cell-mediated immune response that is characterized by the accumulation of T cells and macrophages, release of cytokines and chemokines, and organization of responding cells into granulomas. Clusters of disease in families and communities suggest a genetic predisposition, shared exposures, or, less likely, person-to-person transmission.
The inflammatory process leads to formation of noncaseating granulomas, the pathologic hallmark of sarcoidosis. Granulomas are collections of mononuclear cells and macrophages that differentiate into epithelioid and multinucleated giant cells and are surrounded by lymphocytes, plasma cells, fibroblasts, and collagen. Granulomas occur most commonly in the lungs and lymph nodes but can involve any organ and cause significant dysfunction. Granulomas in the lungs are distributed along lymphatics, with most occurring in peribronchiolar, subpleural, and perilobular regions. Granuloma accumulation distorts architecture in affected organs. Whether granulomas lead directly to fibrosis or run a parallel course is not known.
Hypercalcemia may occur because of increased conversion of vitamin D to the activated form (1,25 hydroxy vitamin D) by macrophages. Hypercalciuria may be present, even in patients with normal serum calcium levels. Nephrolithiasis and nephrocalcinosis may occur, sometimes leading to chronic kidney disease.

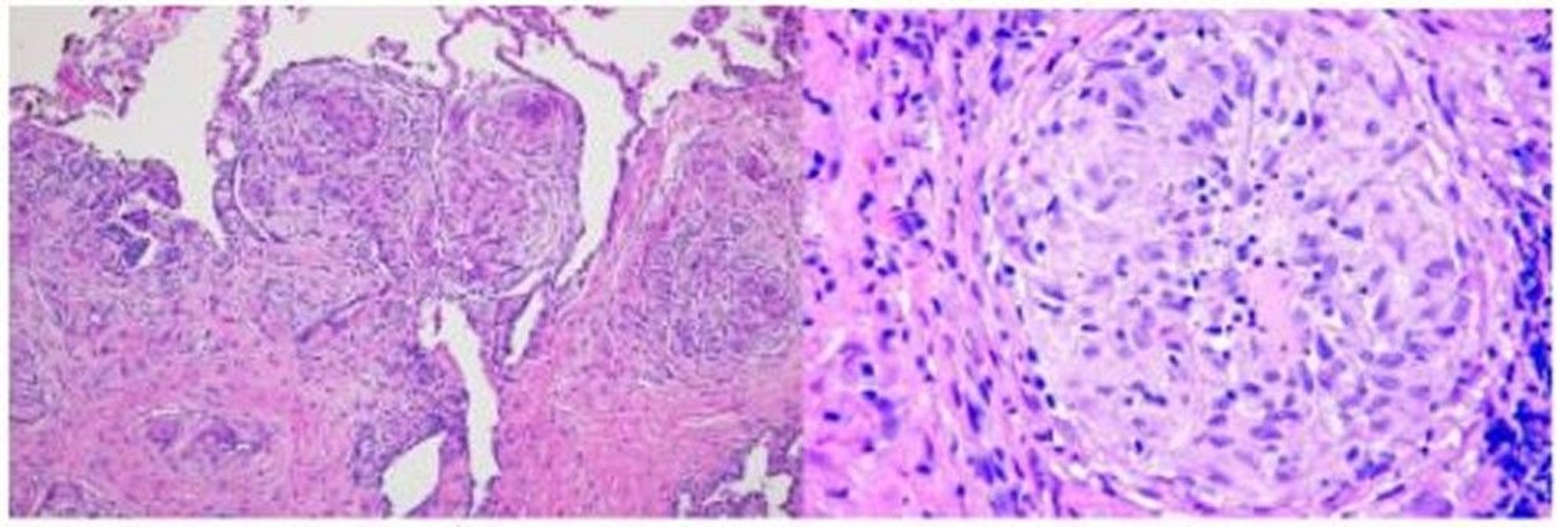
Image courtesy of Birendra P. Sah, MD, FCCP.
Symptoms and Signs of Sarcoidosis
Symptoms and signs depend on the site and degree of involvement and vary over time, ranging from spontaneous remission to chronic indolent illness. Accordingly, frequent reassessment for new symptoms and involvement in different organs is needed. Most cases are probably asymptomatic and thus go undetected. Pulmonary disease occurs in > 90% of adult patients.
Symptoms and signs may include dyspnea, cough, chest discomfort, and crackles. Fatigue, malaise, weakness, anorexia, weight loss, and low-grade fever are also common. Sarcoidosis can manifest as fever of unknown origin. Systemic involvement causes various symptoms (see table Systemic Involvement in Sarcoidosis), which vary by race, sex, and age. Black people are more likely than White people to have involvement of the eyes, liver, bone marrow, peripheral lymph nodes, and skin; erythema nodosum is an exception. Women are more likely to have erythema nodosum and eye or nervous system involvement. Men and older patients are more likely to have hypercalcemia.
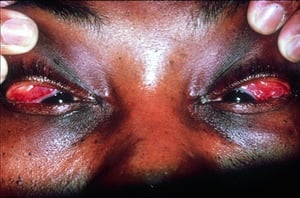
© Springer Science+Business Media
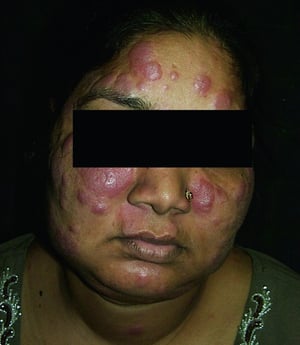
© Springer Science+Business Media
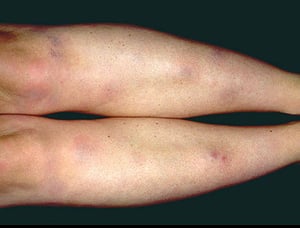
Photo provided by Thomas Habif, MD.
© Springer Science+Business Media
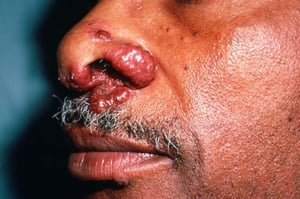
Image courtesy of Karen McKoy, MD.
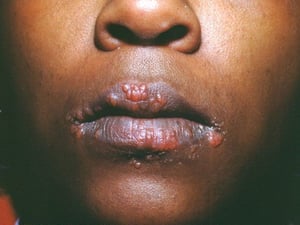
Image courtesy of Karen McKoy, MD.
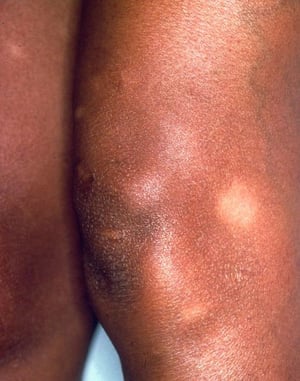
Image courtesy of Karen McKoy, MD.

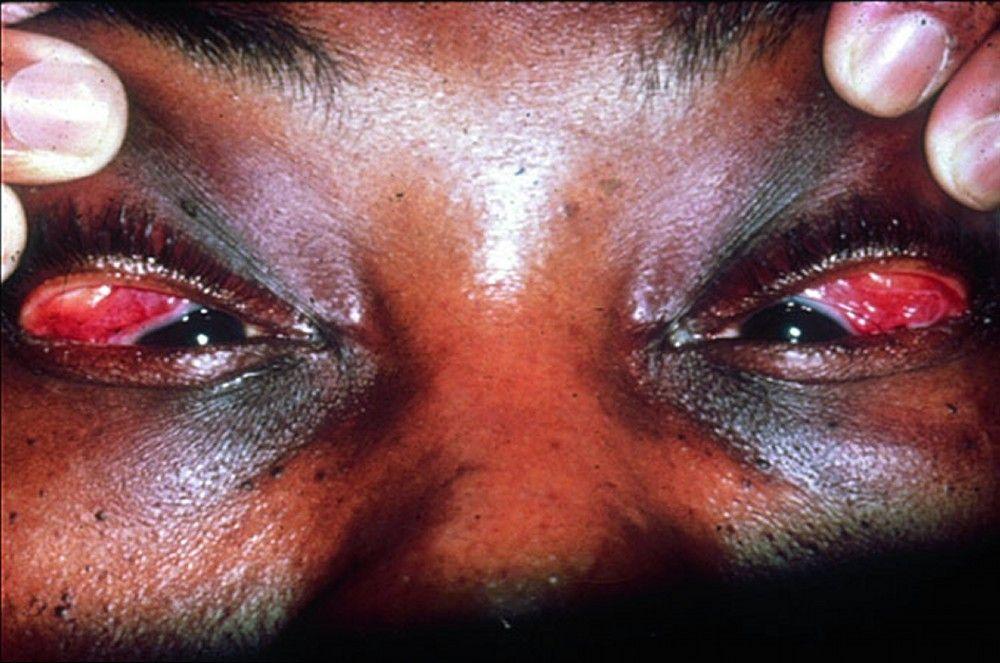
© Springer Science+Business Media

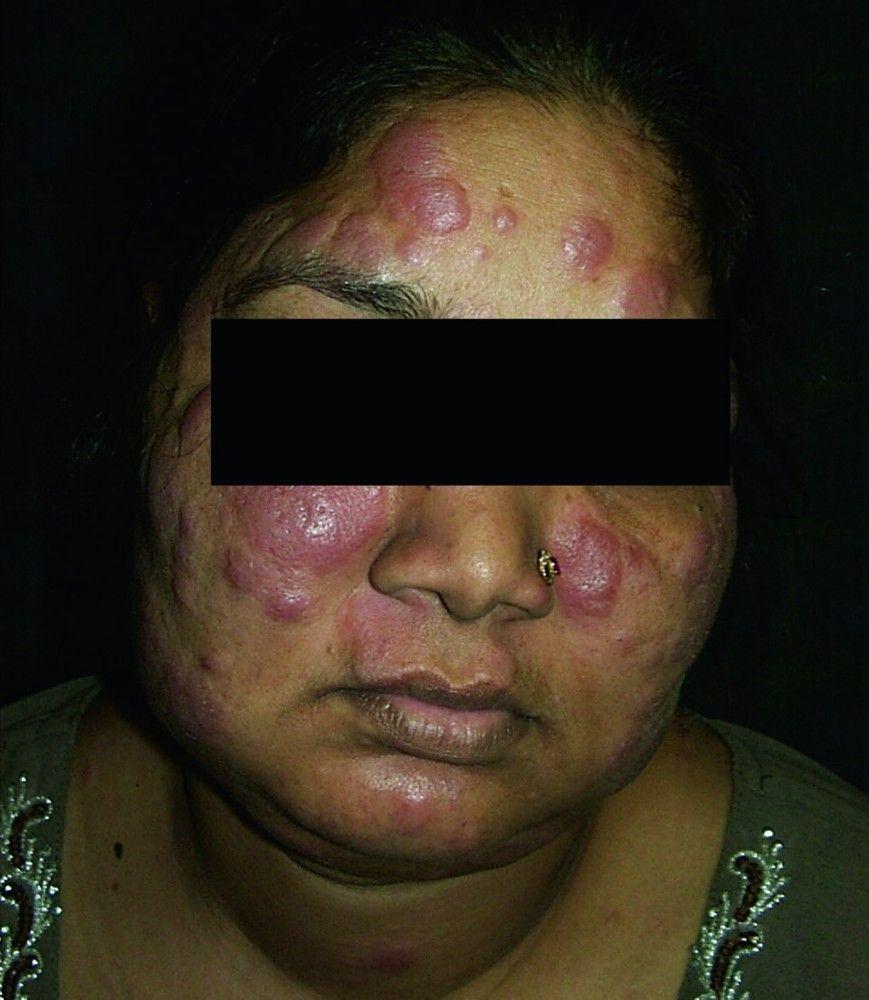
© Springer Science+Business Media

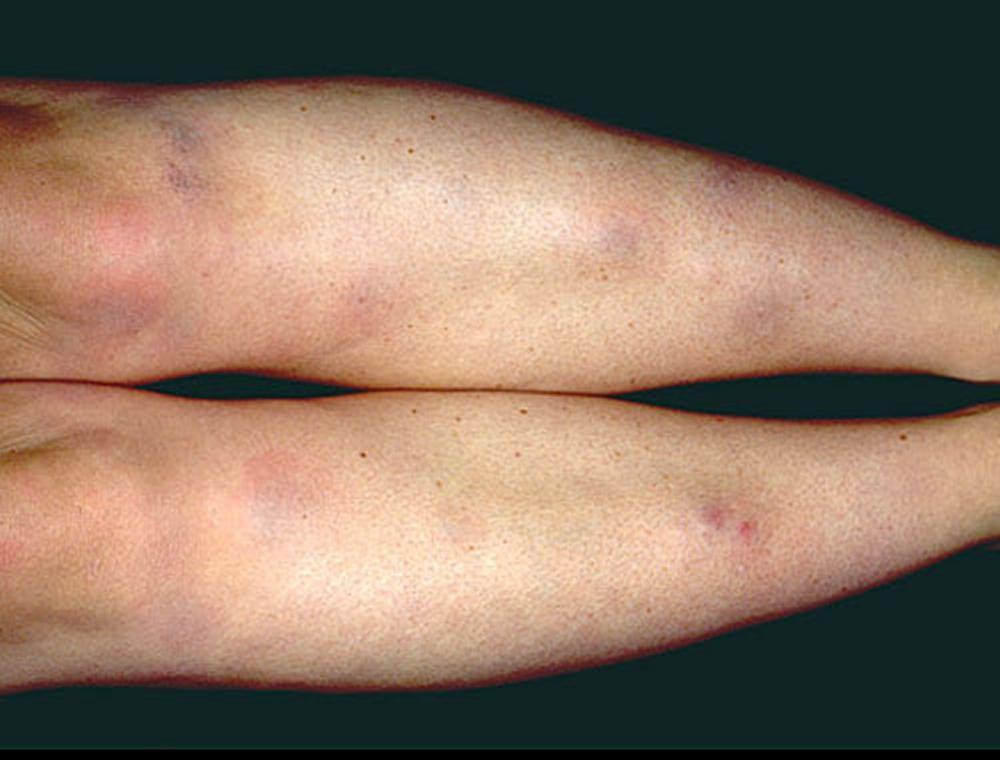
Photo provided by Thomas Habif, MD.

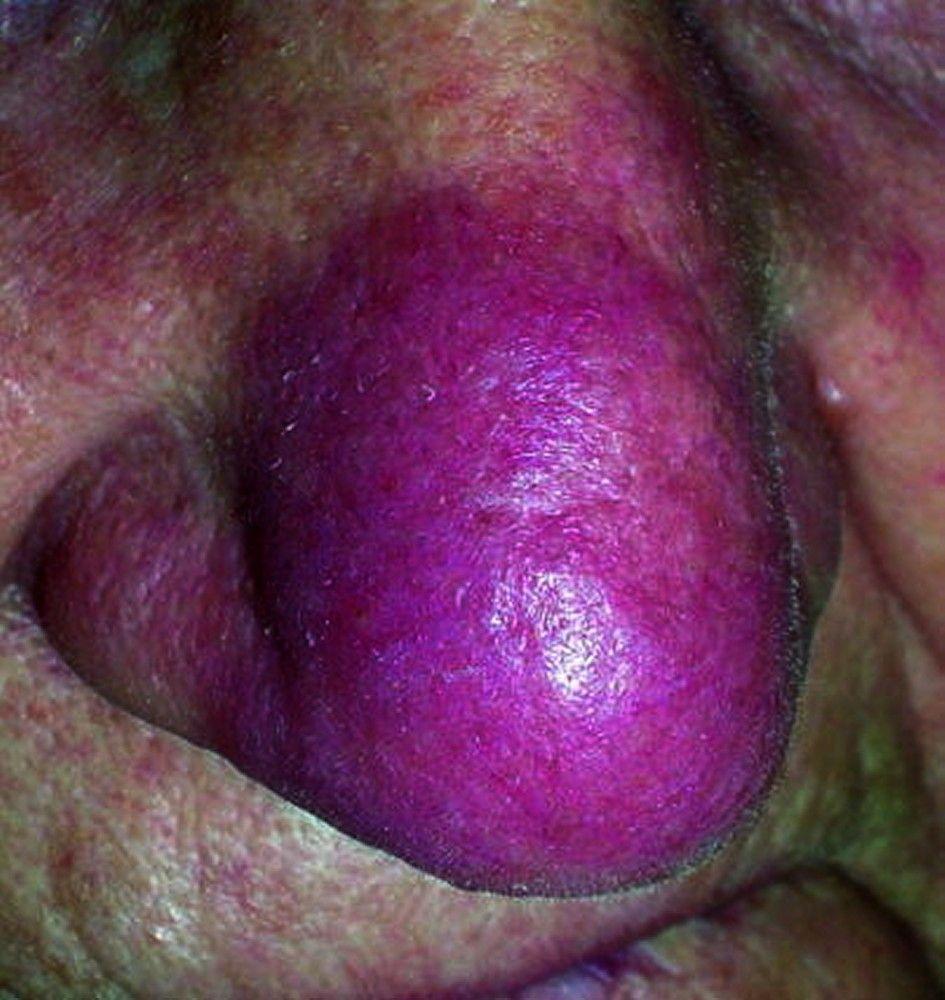
© Springer Science+Business Media

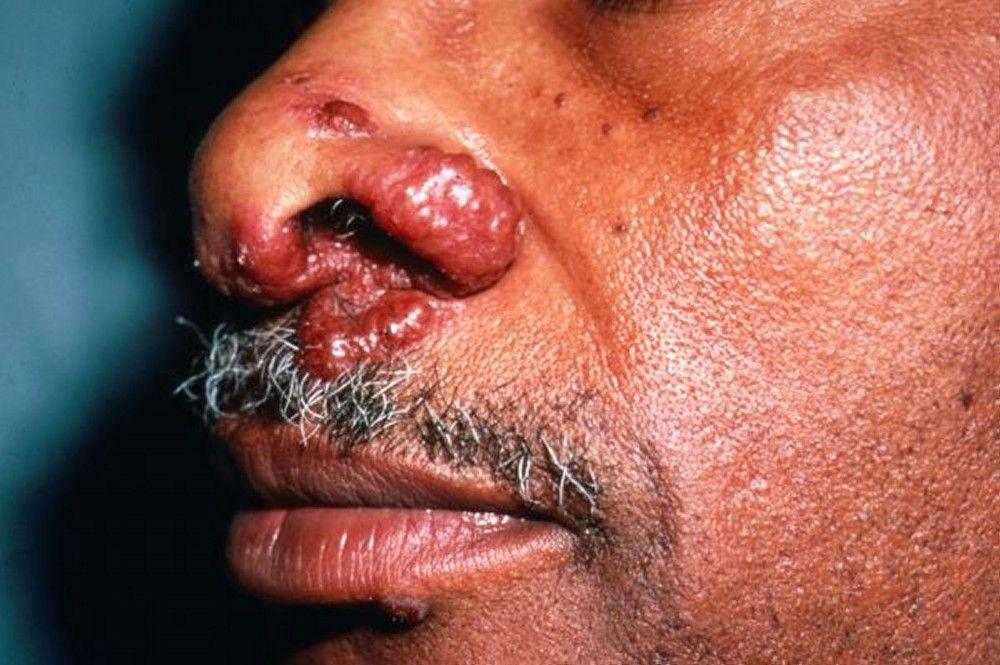
Image courtesy of Karen McKoy, MD.

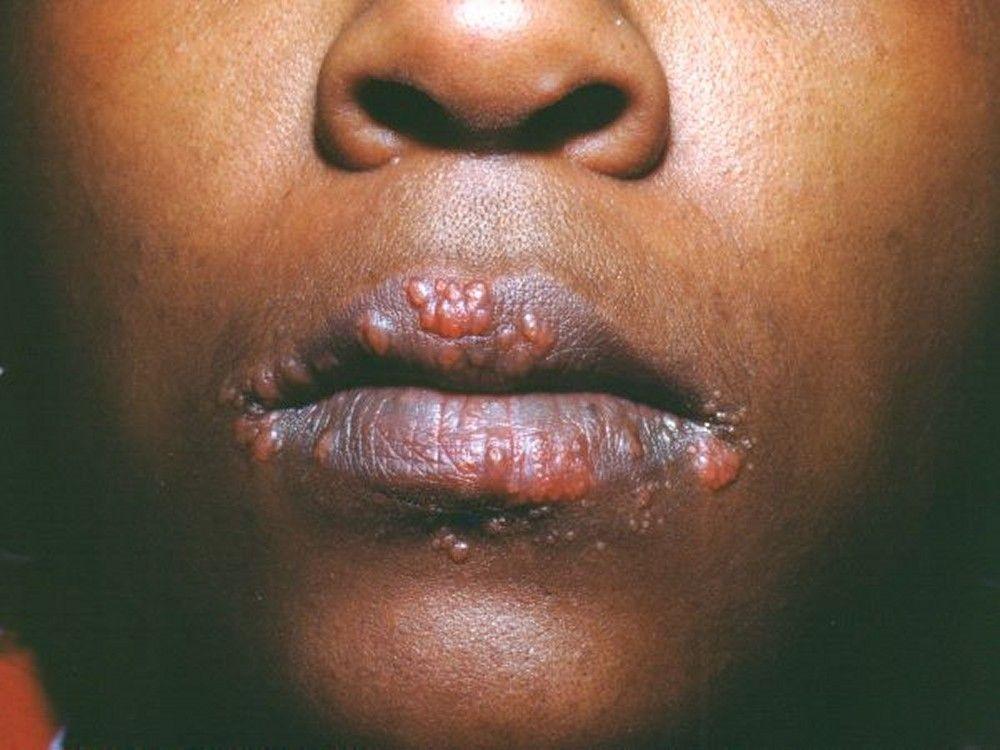
Image courtesy of Karen McKoy, MD.

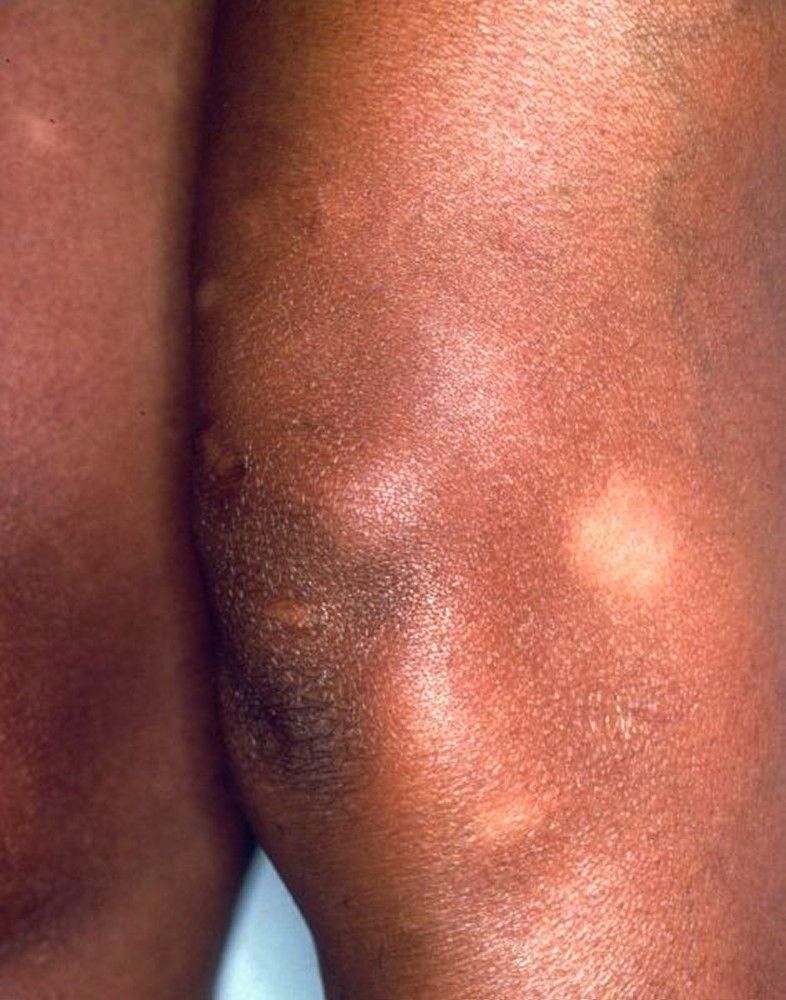
Image courtesy of Karen McKoy, MD.
Children with sarcoidosis may present with Blau syndrome (arthritis, rash, uveitis), or manifestations similar to those of adults. Sarcoidosis may be confused with juvenile idiopathic arthritis (juvenile rheumatoid arthritis) in this age group.
Diagnosis of Sarcoidosis
Chest imaging
Biopsy
Exclusion of other granulomatous disorders
Sarcoidosis is most often suspected when hilar adenopathy, with or without lung infiltrates, is incidentally detected on chest x-ray. Bilateral hilar adenopathy is the most common abnormality.
If sarcoidosis is suspected, a chest x-ray should be the first test if it has not already been done. The x-ray appearance tends to roughly predict the likelihood of spontaneous remission (see table Chest X-ray Staging of Sarcoidosis) in patients with only thoracic lymph node involvement. However, staging sarcoidosis by chest x-ray can be misleading; for example, extrapulmonary sarcoidosis, such as cardiac sarcoidosis or neurologic sarcoidosis, can portend a poor prognosis in the absence of evidence of pulmonary involvement. Also, chest x-rays findings predict pulmonary function poorly, so that chest x-ray appearance may not accurately indicate the severity of pulmonary sarcoidosis.
A normal chest x-ray (stage 0) does not exclude the diagnosis of sarcoidosis, particularly when cardiac or neurologic involvement is suspected. A high-resolution CT is more sensitive for detecting hilar and mediastinal lymphadenopathy and parenchymal abnormalities. Lung parenchymal involvement is predominantly in the upper lobes but can be in any part of lungs. CT findings (see also image Chest CT Scan in Pulmonary Sarcoidosis) in more advanced stages (II to IV) include the following:
Thickening of the bronchovascular bundles and bronchial walls
Beading of the interlobular septa
Ground-glass opacification
Parenchymal nodules, cysts, or cavities
Traction bronchiectasis
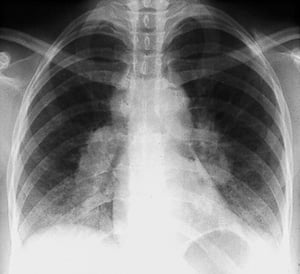
By permission of the publisher. From Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.
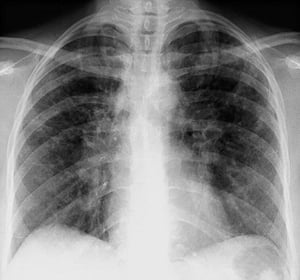
By permission of the publisher. From Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.
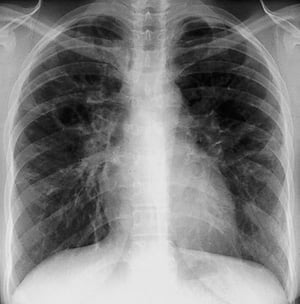
By permission of the publisher. From Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.
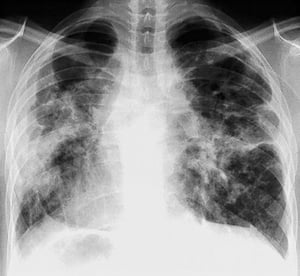
By permission of the publisher. From: Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.
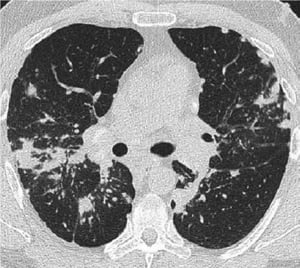
Image courtesy of Birendra P. Sah, MD, FCCP.

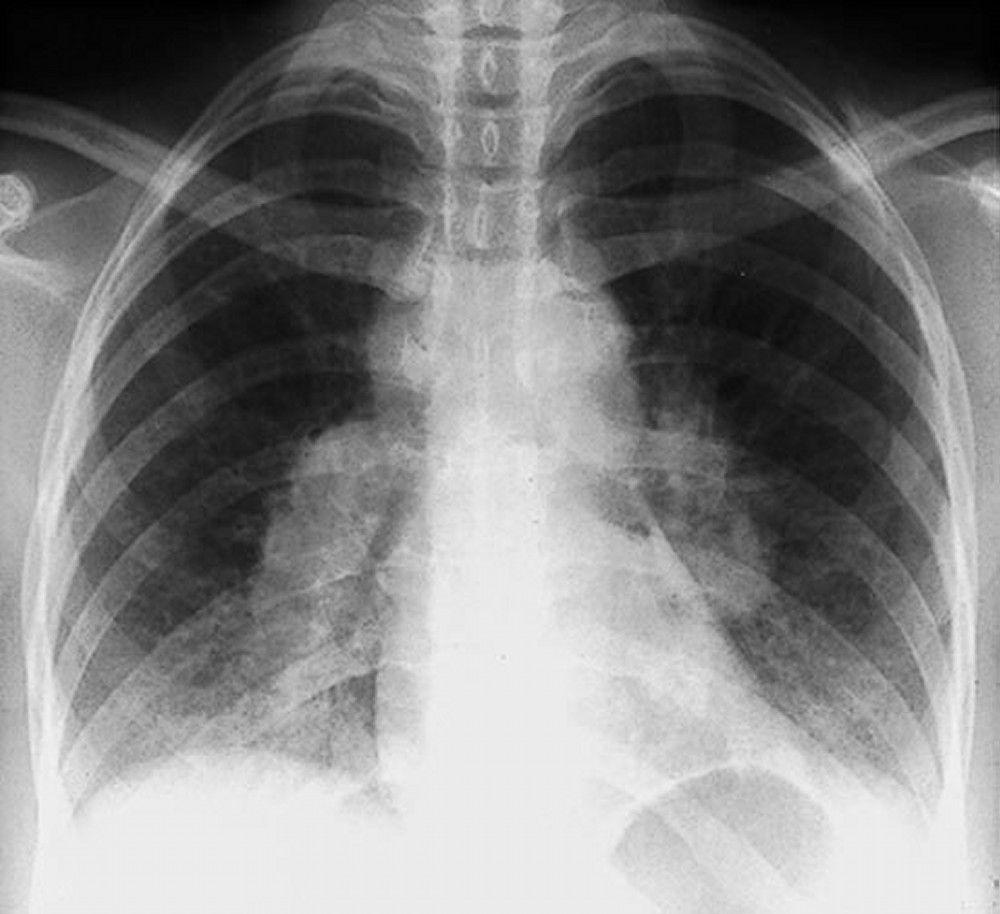
By permission of the publisher. From Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.

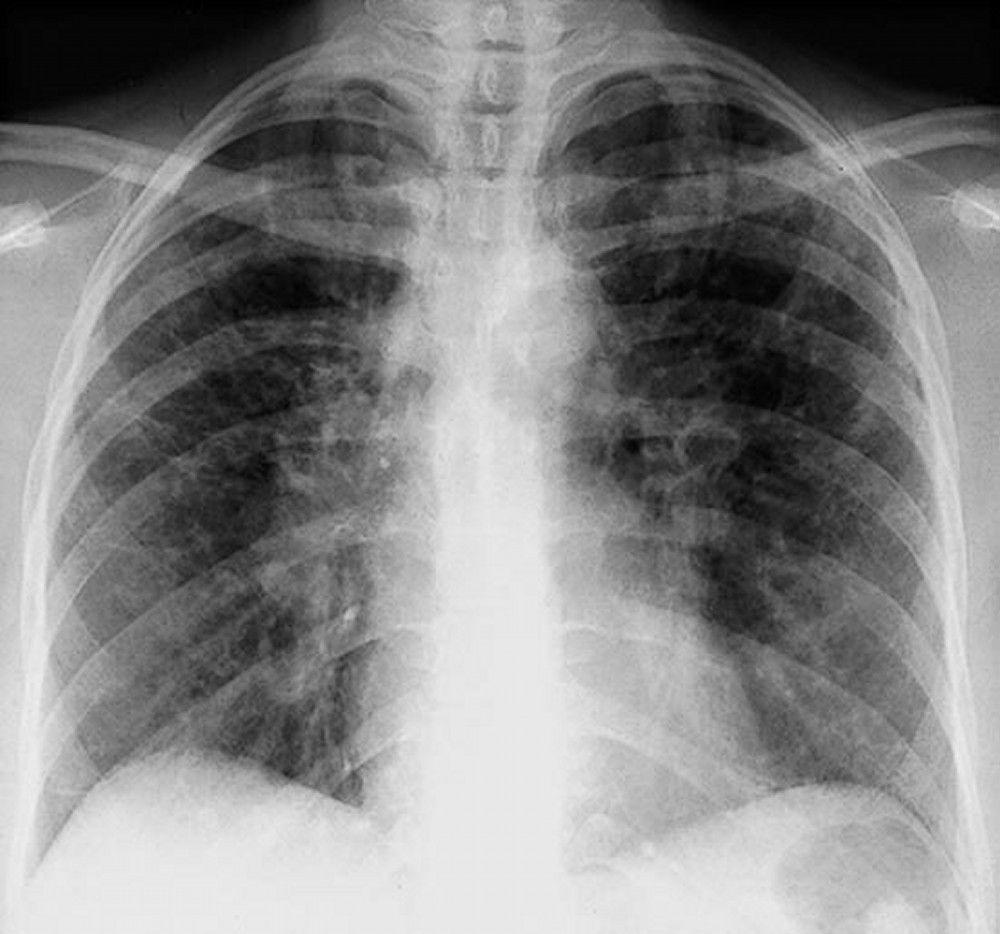
By permission of the publisher. From Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.

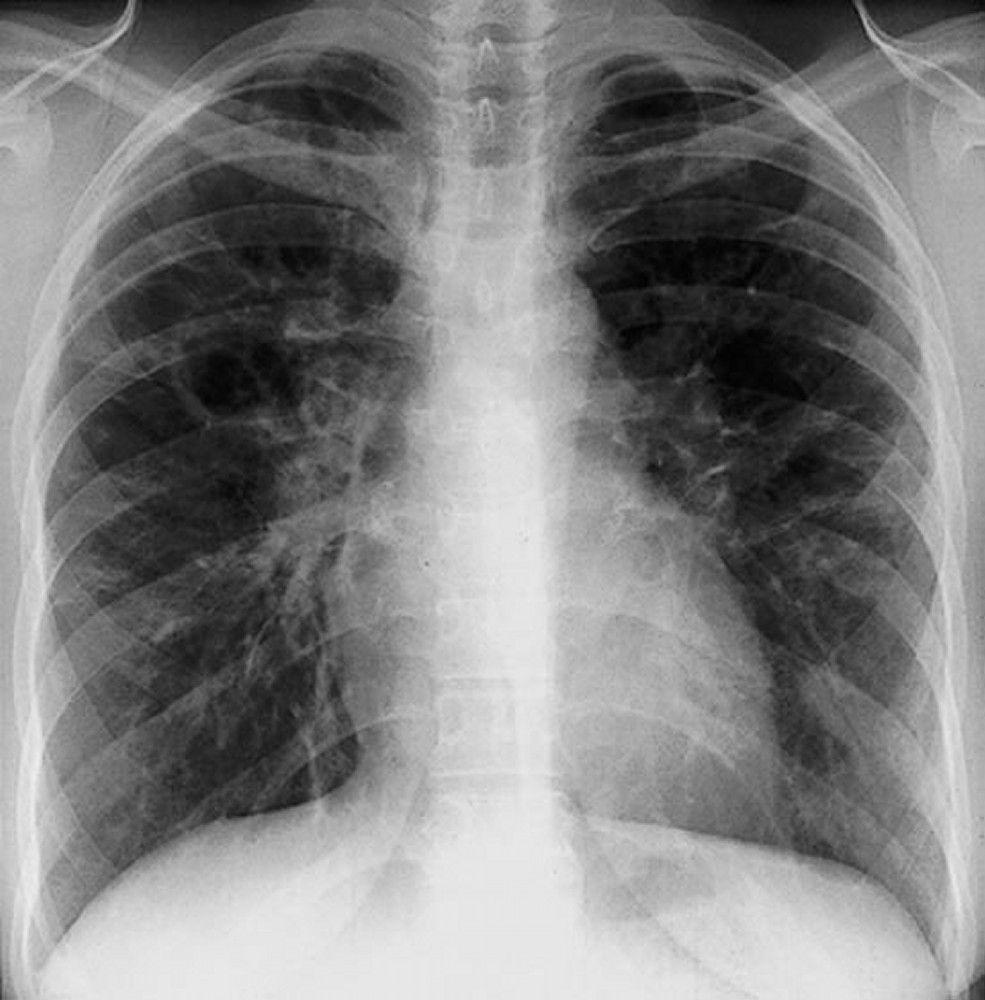
By permission of the publisher. From Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.

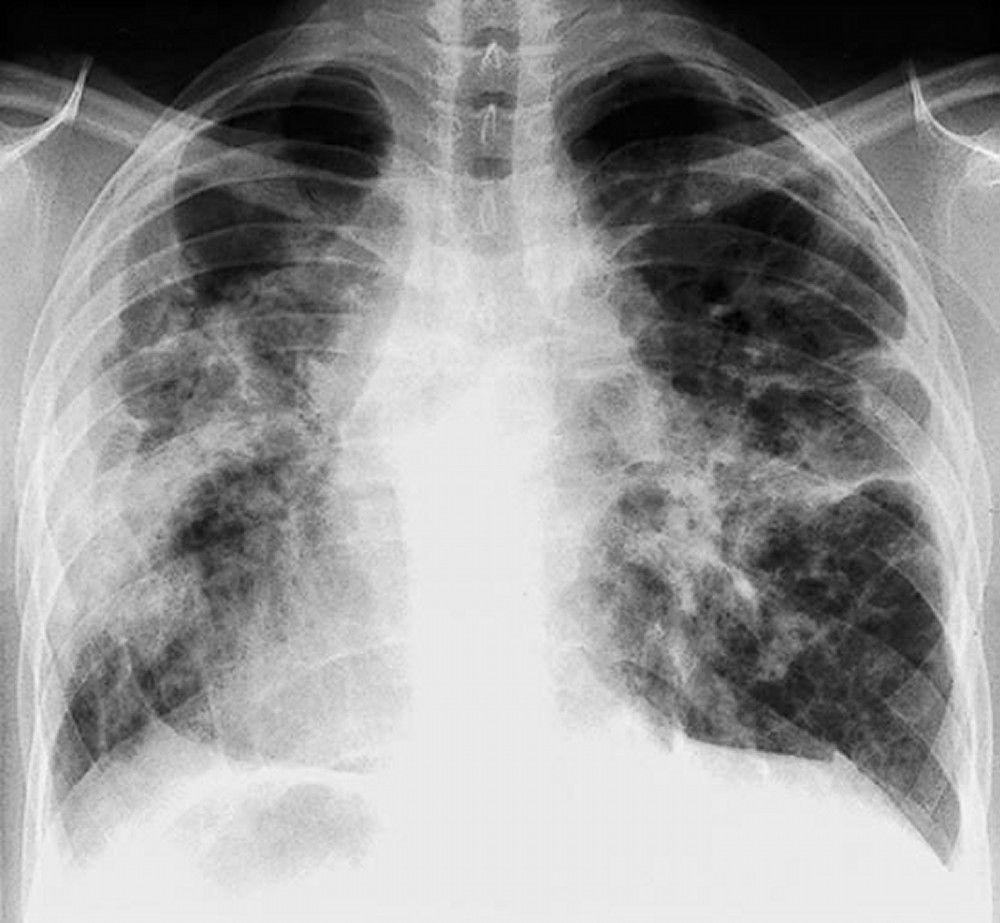
By permission of the publisher. From: Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.

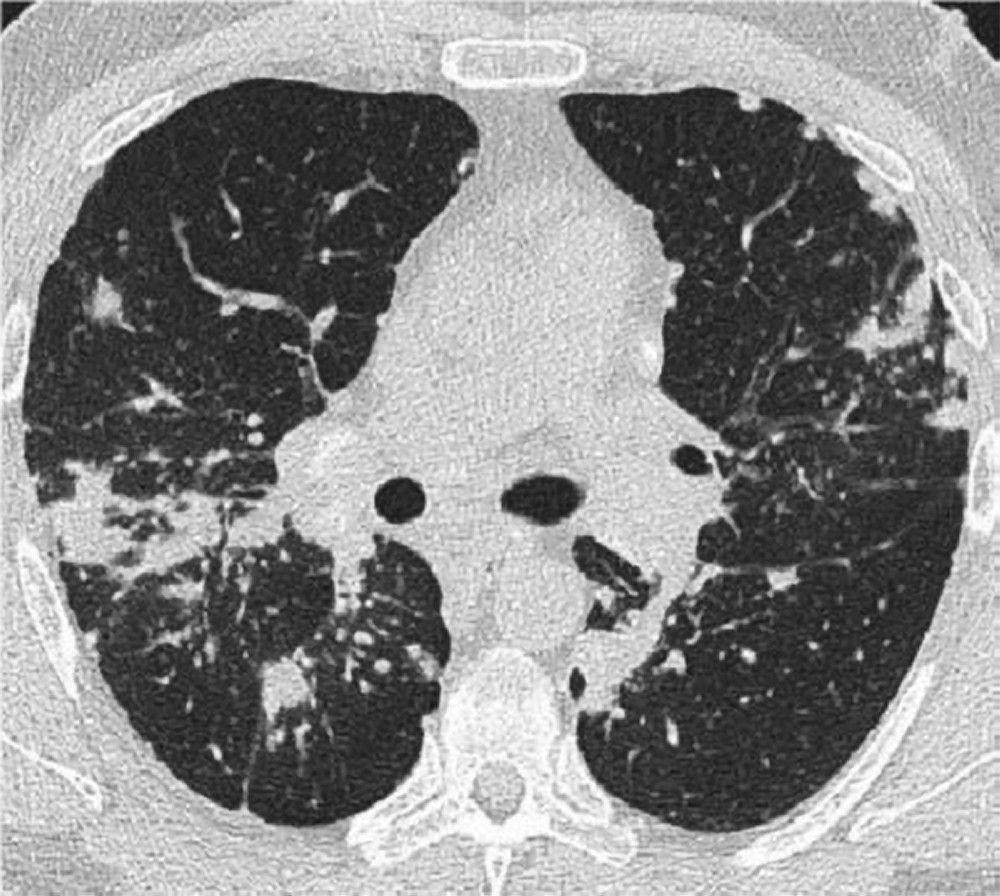
Image courtesy of Birendra P. Sah, MD, FCCP.
When imaging suggests sarcoidosis, the diagnosis is confirmed by demonstration of noncaseating granulomas on biopsy and exclusion of alternative causes of granulomatous disease (see table Differential Diagnosis of Sarcoidosis). Löfgren syndrome does not require confirmation by biopsy.
The diagnostic evaluation, therefore, requires the following:
Selection of a biopsy site
Exclusion of other causes of granulomatous disease
Assessment of the severity and extent of disease to determine whether therapy is indicated
Sites for biopsy
Appropriate biopsy sites may be obvious from physical examination and initial assessment; peripheral lymph nodes, skin lesions, and conjunctivae are all easily accessible. Endobronchial ultrasound–guided transbronchial needle aspiration (EBUS-TBNA) of a mediastinal or hilar lymph node has a reported diagnostic yield of about 90% and is the diagnostic procedure of choice in patients with intrathoracic involvement.
When pulmonary sarcoidosis is suspected and EBUS-TBNA is nondiagnostic, bronchoscopic transbronchial lung biopsy with bronchoalveolar lavage (BAL) can be used. It can also be used in patients without any lung parenchymal infiltrates because diagnostic yield for transbronchial lung biopsy in stage I sarcoidosis is about 50%. If the bronchoscopic transbronchial biopsy is nondiagnostic, it can be tried a second time.
If EBUS-TBNA and bronchoscopic transbronchial biopsies are nondiagnostic or if bronchoscopy cannot be tolerated, mediastinoscopy can be done to biopsy mediastinal or hilar lymph nodes, or video-assisted thoracoscopic (VAT) lung biopsy or open-lung biopsy can be done to obtain lung tissue. If sarcoidosis is strongly suspected but a biopsy site is not evident based on examination or imaging findings, positron emission tomography (PET) scanning can help identify occult active sites such as heart, bone, muscle, and brain.
Exclusion of other diagnoses
Exclusion of other diagnoses is critical, especially when symptoms and x-ray signs are minimal, because many other disorders and processes can cause granulomatous inflammation (see table Differential Diagnosis of Sarcoidosis). Biopsy tissue should be cultured for fungi and mycobacteria. Exposure history to occupational (eg, silica, beryllium), environmental (eg, moldy hay, birds, and other antigenic triggers of hypersensitivity pneumonitis), and infectious (eg, tuberculosis, coccidioidomycosis, histoplasmosis) antigens should be explored. Purified protein derivative (PPD) skin testing or interferon gamma release assay should be done early in the assessment.
Disease severity assessment
Severity is assessed according to organ involvement so, for example, with only pulmonary involvement
Pulmonary function tests
Pulmonary function test results are often normal in early stages but demonstrate restriction and reduced diffusing capacity for carbon monoxide (DLCO) in advanced disease. Airflow obstruction also occurs and may suggest involvement of the bronchial mucosae. Adding a 6-minute walk test may characterize functional impairment more comprehensively than the results of pulmonary function tests alone. Patients with extensive lung involvement may have normal oxygen saturation at rest but may show desaturation with exertion.
Recommended routine screening tests for extrapulmonary disease include
12- lead ECG, Holter monitoring, and echocardiography
Slit-lamp ophthalmologic examination
Routine blood tests to evaluate renal and hepatic function
Serum calcium levels and 24-hour urinary calcium excretion
Imaging
Laboratory testing plays an adjunctive role in establishing the diagnosis and determining the extent of organ involvement. Complete blood count with differential may show anemia, eosinophilia, or leukopenia. Serum calcium should be measured to detect hypercalcemia. Blood urea nitrogen (BUN), creatinine, and liver test results may be elevated in renal and hepatic sarcoidosis. Total protein may be elevated because of hypergammaglobulinemia. Elevated erythrocyte sedimentation rate is common but nonspecific. Measurement of calcium in a urine specimen collected over 24 hours is recommended to exclude hypercalciuria, even in patients with normal serum calcium levels. Elevated serum angiotensin-converting enzyme (ACE) levels can suggest sarcoidosis but are nonspecific and may be elevated in patients with other conditions (eg, hyperthyroidism, diabetes, Gaucher disease, silicosis, mycobacterial disease, fungal infections, hypersensitivity pneumonitis, lymphoma). Angiotensin-converting enzyme (ACE) levels, if elevated, may be useful for monitoring adherence with corticosteroid treatment. ACE levels plummet even when patients are taking low-dose corticosteroids.
Bronchoalveolar lavage should be done along with bronchoscopic biopsy to rule out suspected infections (eg, when findings with less invasive means such as imaging are not typical of sarcoidosis) and to exclude other forms of interstitial lung disease if the diagnosis of sarcoidosis is in doubt. The findings on BAL vary considerably, but lymphocytosis (lymphocytes > 15%), a CD4+/CD8+ ratio of > 3.5 in the lavage fluid cell differential, or both suggest the diagnosis in the proper clinical context. However, absence of these findings does not exclude sarcoidosis.
Treatment of Sarcoidosis
Nonsteroidal anti-inflammatory drugs (NSAIDs)
Corticosteroids
Immunosuppressants
Anti-tumor necrosis factor-alpha antibodies
Because sarcoidosis often spontaneously resolves, asymptomatic patients and patients with mild symptoms do not require treatment, although they should be monitored for signs of deterioration. These patients can be followed with serial chest x-rays, pulmonary function tests (including diffusing capacity), and markers of extrathoracic involvement (eg, routine renal and liver function testing, annual slit-lamp ophthalmologic examination). The frequency of follow-up testing is determined by the severity of disease.
Patients who require treatment regardless of chest x-ray stage include those with the following:
Worsening symptoms
Limitation of activity
Markedly abnormal or deteriorating lung functions
Worrisome x-ray changes (eg, cavitation, fibrosis, conglomerate masses, signs of pulmonary hypertension)
Heart, nervous system, or eye involvement
Renal or hepatic failure
Moderate to severe hypercalcemia
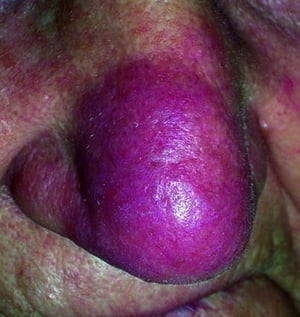
Disfiguring skin (eg, lupus pernio) or joint disease
NSAIDs)are used to treat musculoskeletal discomfort.
Corticosteroids
The optimal duration of treatment is unknown. Premature taper can result in relapse. The drug is slowly stopped if response is absent or equivocal. Corticosteroids can ultimately be stopped in most patients, but because relapse occurs up to 50% of the time, monitoring should be repeated, usually every 3 to 6 months. Corticosteroid treatment should be resumed for recurrence of symptoms and signs. Because angiotensin-converting enzyme (ACE) production is suppressed with low doses of corticosteroids, serial serum ACE levels may be useful in assessing adherence with corticosteroid treatment in patients who have elevated ACE levels.
Inhaled corticosteroids can relieve cough in patients with endobronchial involvement. An inhaled bronchodilator can be added in patients with obstructive airway disease.
Topical corticosteroids may be useful in dermatologic, nasal sinus, and ocular disease.
Prophylaxis against Pneumocystis jirovecii
vitamin D (1, 25 dihydroxy vitamin D) by sarcoidal granulomas. Serum and 24-hour urinary calcium measurements should be normal before starting such supplements.
Pearls & Pitfalls
|
Immunosuppressants
Immunosuppressants are used when
Prior to adding other immunosuppressants, possible reasons for lack of clinical improvement, such as noncompliance, comorbid disease (eg, asthma, heart failure, anemia), pulmonary hypertension, and end-stage fibrosis should be considered.
Relapse is common after stopping an immunosuppressant.
Anti-tumor necrosis factor-alpha antibodies
Other treatment considerations
Patients who have heart block or ventricular arrhythmias due to cardiac involvement should receive an implantable cardiac defibrillator and pacemaker as well as drug therapy.
No available drugs have consistently prevented pulmonary fibrosis.
Treatment of sarcoidosis-associated pulmonary arterial hypertension (SPAH) is supportive with diuresis and supplemental oxygen. The role of pulmonary vasodilators in treating SPAH has not been well established; a few small studies have suggested efficacy, but larger studies are needed to confirm it (1).
Organ transplantation is an option for patients with end-stage pulmonary, cardiac, or liver involvement, although disease may recur in the transplanted organ.
Patients with sarcoidosis and moderate or severe impairment of pulmonary or cardiac function are at increased risk of developing severe forms of COVID-19 infection and mortality. As in patients with other inflammatory lung diseases during the COVID-19 pandemic, immunosuppressants should be used judiciously. SARS-CoV-2 vaccination is strongly recommended in patients with sarcoidosis because it could reduce the mortality and severity of illness due to COVID-19 infection. An ongoing clinical trial aims to better assess the efficacy of SARS-CoV-2 vaccination in patients with sarcoidosis and in patients taking immunosuppressants.
Treatment reference
1. Humbert M, Kovacs G, Hoeper MM, et al: 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 61(1): 1-144, 2023. doi:10.1183/13993003.00879-2022
Prognosis for Sarcoidosis
Although spontaneous remission is common, disease manifestations and severity are highly variable, and many patients require corticosteroids to relieve symptoms or slow progressive organ function decline at some time during the disease course. Thus, serial monitoring for evidence of relapse is imperative. Almost two thirds of patients with sarcoidosis eventually achieve remission with few or no sequelae. In about 50% of patients who have spontaneous remission, remission occurs within the first 3 years after diagnosis. Fewer than 10% of these patients relapse after 2 years. Patients who do not experience remission within 2 to 3 years are likely to have chronic disease.
Sarcoidosis is thought to be chronic in up to 30% of patients, and 10 to 20% experience permanent sequelae. The disease is fatal in 1 to 5% of patients, typically due to respiratory failure caused by pulmonary fibrosis, and less often due to pulmonary hemorrhage caused by aspergilloma. However, in Japan, infiltrative cardiomyopathy causing arrhythmias and heart failure is the most common cause of death.
Prognosis is worse for patients with extrapulmonary sarcoidosis and for Black people. Remission occurs in 89% of White patients and 76% of Black patients with no extrathoracic disease and in 70% of White patients and 46% of Black patients with extrathoracic disease.
Good prognostic signs include
Löfgren syndrome (triad of acute polyarthritis, erythema nodosum, and hilar adenopathy)
Poor prognostic signs include
Chronic uveitis
Lupus pernio
Chronic hypercalcemia
Neurosarcoidosis
Cardiac involvement
Extensive pulmonary involvement and/or development of pulmonary hypertension
Little difference is demonstrable in long-term outcome between treated and untreated patients, and relapse is common when treatment ends.
Key Points
Systemic and extrapulmonary involvement is common with sarcoidosis, but > 90% of adult patients have pulmonary involvement.
Obtain a chest imaging study but confirm the diagnosis by biopsy, usually endobronchial ultrasound-guided transbronchial needle aspiration of a mediastinal or hilar lymph node.
Assess pulmonary severity with pulmonary function testing and exercise pulse oximetry.
Test for extrapulmonary involvement with ECG, slit-lamp examination, renal and hepatic function tests, and serum and urinary calcium testing.
Treat patients with systemic corticosteroids when indicated (eg, severe symptoms, hypercalcemia, progressive decline in organ function, cardiac or neurologic involvement).
Treat with immunosuppressants if patients cannot tolerate moderate doses of corticosteroids, sarcoidosis is resistant to corticosteroids, or if corticosteroids are required long term.




