

Cerebral palsy refers to a group of nonprogressive conditions characterized by impairments in muscle tone, voluntary movement, and/or posture, likely resulting from prenatal developmental malformations or perinatal or postnatal central nervous system damage. Cerebral palsy manifests before age 2 years. Diagnosis is by history and physical examination. Management may include physical and occupational therapies, braces, medications, botulinum toxin injections, intrathecal baclofen, or orthopedic surgery.Cerebral palsy refers to a group of nonprogressive conditions characterized by impairments in muscle tone, voluntary movement, and/or posture, likely resulting from prenatal developmental malformations or perinatal or postnatal central nervous system damage. Cerebral palsy manifests before age 2 years. Diagnosis is by history and physical examination. Management may include physical and occupational therapies, braces, medications, botulinum toxin injections, intrathecal baclofen, or orthopedic surgery.
Cerebral palsy (CP) is a term that describes a group of heterogenous conditions that causes nonprogressive spasticity, ataxia, or involuntary movements; it is not a specific disease (1). Similar clinical conditions that manifest after 2 years old are not considered cerebral palsy.
The global prevalence of CP is 2/1000 live births (2). Two risk factors for increased prevalence are low birth weight and preterm birth: neonates weighing between 1000 to 1499 g (59.18/1000 live births) and preterm infants < 28 weeks of gestation (111.8/1000 live births).

General references
1. Panda S, Singh A, Kato H, Kokhanov A. Cerebral Palsy: A Current Perspective. Neoreviews. 2024;25(6):e350-e360. doi:10.1542/neo.25-6-e350
2. Oskoui M, Coutinho F, Dykeman J, Jetté N, Pringsheim T. An update on the prevalence of cerebral palsy: a systematic review and meta-analysis [published correction appears in Dev Med Child Neurol. 2016 Mar;58(3):316. doi: 10.1111/dmcn.12662]. Dev Med Child Neurol. 2013;55(6):509-519. doi:10.1111/dmcn.12080
Etiology of Cerebral Palsy
The etiology of cerebral palsy is multifactorial, and a specific cause is sometimes hard to establish. Risk factors that often contribute include prematurity and the disorders it is often associated with, such as in utero disorders, neonatal encephalopathy, and/or chronic bilirubin encephalopathy (formerly known as kernicterus). Perinatal factors (eg, perinatal asphyxia, stroke, central nervous system [CNS] infections) cause a significant minority of cases.
Abnormal whole exome sequencing or whole genome sequencing results can be seen in more than 30% of patients with CP (1).
CNS trauma or a severe systemic disorder (eg, stroke, meningitis, sepsis, dehydration) during early childhood (before 2 years of age) may also cause CP.
Examples of types of CP are
Spastic diplegia after preterm birth
Spastic hemiplegia in term infants with neonatal stroke
Spastic quadriplegia after perinatal asphyxia in preterm infants, small-for-gestational-age infants, or both
Dyskinetic types, including athetoid and dystonic forms after perinatal asphyxia or chronic bilirubin encephalopathy
Ataxic types, most commonly with unknown etiologies, sometimes due to genetic cerebellar disorders
Etiology reference
1. Moreno-De-Luca A, Millan F, Pesacreta DR, et al. Molecular Diagnostic Yield of Exome Sequencing in Patients With Cerebral Palsy. JAMA. 2021;325(5):467-475. doi:10.1001/jama.2020.26148
Symptoms and Signs of Cerebral Palsy
Clinical manifestation for all types of CP usually includes lagging motor development and often persistent infantile reflex patterns, hyperreflexia, and altered muscle tone.
CP may have associated impairments and conditions such as chronic pain, speech and language disorders (with or without hearing impairment), seizures, behavioral problems, hip displacement, and drooling (3). Strabismus and other visual defects may occur. Children with athetosis due to chronic bilirubin encephalopathy commonly have nerve deafness and upward gaze paralysis.
Many children with spastic hemiplegia or diplegia have normal intelligence; on the other hand, children with spastic quadriplegia or mixed CP may have severe intellectual disability.
Types of cerebral palsy
CP is categorized mainly as one of the following, depending on which parts of the CNS are malformed or damaged (1, 2):
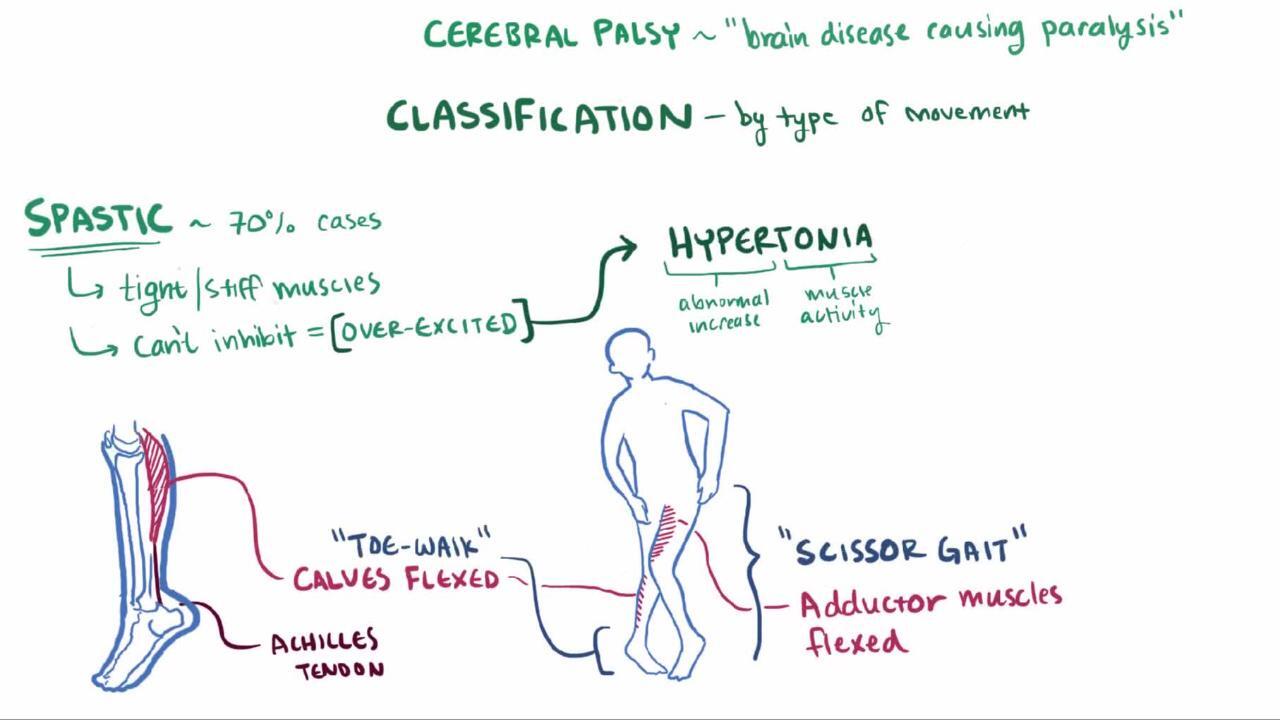
Spastic CP is the most common type and occurs in > 80% of cases (2). Spastic CP may manifest as spastic hemiplegia, quadriplegia, or diplegia. Spasticity is a state of resistance to passive range of motion; resistance increases with increasing speed of that motion. It is due to upper motor neuron involvement and may mildly or severely affect motor function. Usually, deep tendon reflexes in affected limbs are increased, muscles are hypertonic, and voluntary movements are weak and poorly coordinated. Joint contractures develop, and joints may become misaligned. A scissoring gait and toe walking are typical. In mild cases, impairment may occur only during certain activities (eg, running). Corticobulbar impairment of oral, lingual, and palatal movement, with consequent dysarthria or dysphagia, commonly occurs with quadriplegia.
Dyskinetic CP (athetoid CP or dystonic CP) is the second most common type. It occurs in approximately 15% of people with CP and results from basal ganglia involvement. Athetoid or dystonic CP is defined by slow, writhing, involuntary movements of the proximal extremities and trunk (athetoid movements), often activated by attempts at voluntary movement or by excitement. Abrupt, jerky, distal (choreic) movements may also occur. Movements increase with emotional tension and disappear during sleep. Dysarthria occurs and is often severe.
Ataxic CP is rare and results from involvement of the cerebellum or its pathways. Weakness, incoordination, and intention tremor cause unsteadiness, a wide-based gait, and difficulty with rapid or fine movements.
Mixed CP is common. This type manifests most often with spasticity and athetosis/dystonia.
A tool called the Gross Motor Function Classification System–Expanded and Revised (GMFCS–E&R) can be used to describe the gross motor function of children with CP. The system categorizes gross motor function into 5 different groups. It provides a description of current motor function that helps identify current and future needs for mobility aids.
Symptoms and signs references
1. Panda S, Singh A, Kato H, Kokhanov A. Cerebral Palsy: A Current Perspective. Neoreviews. 2024;25(6):e350-e360. doi:10.1542/neo.25-6-e350
2. Monbaliu E, Himmelmann K, Lin JP, et al. Clinical presentation and management of dyskinetic cerebral palsy. Lancet Neurol. 2017;16(9):741–749. doi:10.1016/S1474-4422(17)30252-1
3. Novak I, Hines M, Goldsmith S, Barclay R. Clinical prognostic messages from a systematic review on cerebral palsy. Pediatrics. 2012;130(5):e1285-e1312. doi:10.1542/peds.2012-0924
Diagnosis of Cerebral Palsy
History and physical examination
Brain MRI
Testing to exclude hereditary metabolic or neurologic disorders
If CP is suspected, identifying the underlying disorder, if possible, is important. The medical history may suggest a cause. A brain MRI detects abnormalities in most cases and is the preferred imaging modality; CT also may be used. Cranial ultrasound can sometimes be helpful to identify bleeding and leukomalacia.
Historically, the diagnosis of CP was typically made between 12 months and 24 months of age. However, CP can be diagnosed as early as < 5 months corrected age for infants who have detectable risks at birth (eg, prematurity, encephalopathy). The diagnosis can be made using a combination of neuroimaging (brain MRI), standardized neurologic examination (eg, Hammersmith Infant Neurological Examination) (1), and standardized motor assessment (eg, Prechtl Qualitative Assessment of General Movements) (2).
High-risk children should be followed closely and include children with the following:
History of asphyxia or evidence of stroke and periventricular abnormalities (such as periventricular leukomalacia) on cranial ultrasound in preterm infants
Jaundice
Meningitis
Neonatal seizures
Hypertonia
Hypotonia
Reflex suppression
About half of all children with CP have concomitant features of epilepsy. In these patients, an electroencephalogram may be helpful for further management of seizures.
Screening should be performed for visual and hearing defects.
Differential diagnosis
CP should be differentiated from progressive hereditary neurologic disorders and disorders requiring surgical or other specific neurologic treatments. Inborn errors of metabolism should be ruled out via newborn screening or via additional genetic or metabolic tests. (See also Approach to the Patient With a Suspected Inherited Disorder of Metabolism for information regarding specific tests.)
When history and/or brain MRI does not clearly identify a cause, laboratory tests should be done to exclude certain progressive storage disorders that involve the motor system (eg, Tay-Sachs disease, metachromatic leukodystrophy, mucopolysaccharidoses) and metabolic disorders (eg, organic or amino acid metabolism disorders).
Other progressive disorders (eg, infantile neuroaxonal dystrophy) may be suggested by nerve conduction studies and electromyography. These and many other brain disorders that cause CP (and other manifestations) are being increasingly identified with genetic testing (eg, microarray analysis, CP spectrum disorders gene panel, whole exome sequencing analysis, next-generation sequencing technologies), which may be done to check for a specific disorder or to screen for many disorders.
Ataxic CP is particularly hard to distinguish, and, in many children with persistent ataxia, a progressive cerebellar degenerative disorder is ultimately identified as the cause.
Athetosis, self-mutilation, and hyperuricemia in boys is highly suggestive of Lesch-Nyhan syndrome.
Cutaneous or ocular abnormalities may indicate tuberous sclerosis complex, neurofibromatosis, ataxia-telangiectasia, von Hippel–Lindau disease, or Sturge-Weber syndrome.
Infantile spinal muscular atrophy, muscular dystrophies, and neuromuscular junction disorders associated with hypotonia and hyporeflexia usually lack signs of cerebral disease. (See also Spinal Muscular Atrophy (SMAs) and Introduction to Inherited Muscular Disorders.)
Adrenoleukodystrophy begins later in childhood, but other leukodystrophies begin earlier and may be initially mistaken for CP.
Diagnosis references
1. Ljungblad UW, Paulsen H, Tangeraas T, Evensen KAI. Reference Material for Hammersmith Infant Neurologic Examination Scores Based on Healthy, Term Infants Age 3-7 Months. J Pediatr. 2022;244:79-85.e12. doi:10.1016/j.jpeds.2022.01.032
2. Einspieler C, Prechtl HF. Prechtl's assessment of general movements: a diagnostic tool for the functional assessment of the young nervous system. Dev Disabil Res Rev. 2005;11(1):61-67. doi:10.1002/mrdd.20051
Treatment of Cerebral Palsy
Physical and occupational therapies
Sometimes braces, constraint therapy
Sometimes muscle relaxants for spasticity
Botulinum toxin injections to treat spasticity
Intrathecal baclofen to treat spasticity
Assistive devices
Sometimes orthopedic surgery
Physical therapy and occupational therapy for stretching, strengthening, and facilitating effective movement patterns are usually first-line therapy and are often combined with other treatments. Bracing, constraint therapy, and medications may be added as needed.
Botulinum toxin may be injected into muscles to decrease their uneven pull at joints and to prevent fixed contractures for some children with features of spasticity (1). This medication is best used for localized/segmental spasticity in the upper and lower extremities of children with CP.
Muscle relaxants, including baclofen, benzodiazepines (eg, diazepam), tizanidine, and rarely dantrolene, may diminish spasticity. These oral medications are considered first-line treatment for spasticity particularly in children with generalized spasticity. including baclofen, benzodiazepines (eg, diazepam), tizanidine, and rarely dantrolene, may diminish spasticity. These oral medications are considered first-line treatment for spasticity particularly in children with generalized spasticity.
Intrathecal baclofen (via subcutaneous pump and catheter) can be considered for children with severe spasticity who did not respond to a trial of or had significant adverse effects with oral antispasticity medications.
Orthopedic surgery (eg, muscle-tendon release or transfer) may help reduce restricted joint motion or misalignment in children with severe contractures or extreme mobility limitation. Selective dorsal rhizotomy, done by a neurosurgeon, may help a few children if spasticity affects primarily the legs and if cognitive abilities are well-developed.
Assistive devices may increase mobility and communication, help maintain range of motion, and help with activities of daily living in some severely affected children. Children without severe intellectual limitations may take part in adapted exercise programs and even competition. Speech training or other forms of facilitated communication may be needed to enhance interactions.
When intellectual limitations are not severe, children may attend mainstream classes. Training in activities of daily living (eg, washing, dressing, feeding) increases their independence and self-esteem and greatly reduces the burden for family members or other caregivers. Some children require varying degrees of lifelong supervision and assistance. Many children's facilities are establishing transition programs for patients as they become adults and have fewer supports to help with special needs.
Parents of a child with chronic limitations need assistance and guidance in understanding the child’s status and potential and in dealing with their own possible feelings of guilt, anger, denial, and/or sadness (see Effects on the family). These children reach their maximal potential with stable, consistent care and the assistance of public and private agencies (eg, community health agencies, vocational rehabilitation organizations, health organizations such as United Cerebral Palsy).
Treatment references
1. Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society, Delgado MR, Hirtz D, et al. Practice parameter: pharmacologic treatment of spasticity in children and adolescents with cerebral palsy (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2010;74(4):336-343. doi:10.1212/WNL.0b013e3181cbcd2f
2. Novak I, Morgan C, Fahey M, et al. State of the Evidence Traffic Lights 2019: Systematic Review of Interventions for Preventing and Treating Children with Cerebral Palsy. Curr Neurol Neurosci Rep. 2020;20(2):3. Published 2020 Feb 21. doi:10.1007/s11910-020-1022-z
Prognosis for Cerebral Palsy
Most children survive to adulthood. Severe limitations in sucking and swallowing, which may require feeding by gastrostomy tube, can decrease life expectancy because of the increased risk of aspiration pneumonia and respiratory failure (1). Severe motor impairment and intellectual disability can also increase the risk of mortality (2).
Mortality rates have been decreasing worldwide because of advances in treatment and care (1). The goal is for children to develop maximal independence within the limits of their motor and associated deficits. With appropriate management, many children, especially those with spastic diplegia or hemiplegia, can lead near-typical lives.
Prognosis references
1. Blair E, Langdon K, McIntyre S, Lawrence D, Watson L. Survival and mortality in cerebral palsy: observations to the sixth decade from a data linkage study of a total population register and National Death Index. BMC Neurol. 2019;19(1):111. Published 2019 Jun 4. doi:10.1186/s12883-019-1343-1
2. Himmelmann K, Sundh V. Survival with cerebral palsy over five decades in western Sweden. Dev Med Child Neurol. 2015;57(8):762-767. doi:10.1111/dmcn.12718
Prevention of Cerebral Palsy
Antenatally, preventive options include administration of corticosteroids and magnesium sulfate (1), both of which are now standards of care before delivery of very preterm infants.
Postnatally, caffeine and therapeutic hypothermia (ie, cooling) have both become common practice in neonatal intensive care units for neuroprotection in infants with neonatal encephalopathy and asphyxia (Postnatally, caffeine and therapeutic hypothermia (ie, cooling) have both become common practice in neonatal intensive care units for neuroprotection in infants with neonatal encephalopathy and asphyxia (2, 3).
Prevention references
1. Committee Opinion No. 455: Magnesium sulfate before anticipated preterm birth for neuroprotection. Obstet Gynecol. 2010;115(3):669-671. doi:10.1097/AOG.0b013e3181d4ffa5
2. Schmidt B, Roberts RS, Davis P, et al. Caffeine therapy for apnea of prematurity. N Engl J Med. 2006;354(20):2112-2121. doi:10.1056/NEJMoa054065
3. Novak I, Morgan C, Fahey M, et al. State of the Evidence Traffic Lights 2019: Systematic Review of Interventions for Preventing and Treating Children with Cerebral Palsy. Curr Neurol Neurosci Rep. 2020;20(2):3. Published 2020 Feb 21. doi:10.1007/s11910-020-1022-z
Key Points
Cerebral palsy (CP) is a group of conditions (not a specific disorder) that involve nonprogressive spasticity, ataxia, and/or involuntary movements.
Etiology is often multifactorial and involves prenatal and perinatal factors that are associated with central nervous system (CNS) malformation or damage (eg, genetic and in utero disorders, prematurity, chronic bilirubin encephalopathy, perinatal asphyxia, stroke, CNS infections).
Intellectual disability and other neurologic and systemic conditions (eg, strabismus, deafness) may accompany the motor impairments of CP.
Symptoms manifest before 2 years of age; later onset of similar symptoms suggests another neurologic disorder.
Do brain MRI and, if needed, testing for hereditary or genetic, metabolic, and neurologic disorders.
Treatment depends on the nature and degree of disability, but physical therapy and occupational therapy are typically used; some children benefit from bracing, botulinum toxin, benzodiazepines, other muscle relaxants, intrathecal baclofen, and/or surgery (eg, muscle-tendon release or transfer, rarely dorsal rhizotomy).Treatment depends on the nature and degree of disability, but physical therapy and occupational therapy are typically used; some children benefit from bracing, botulinum toxin, benzodiazepines, other muscle relaxants, intrathecal baclofen, and/or surgery (eg, muscle-tendon release or transfer, rarely dorsal rhizotomy).
Drug Information for the Topic





