

Lymphedema is edema of a limb due to lymphatic hypoplasia (primary) or to obstruction or disruption (secondary) of lymphatic vessels. Symptoms and signs, when chronic, are brawny, fibrous, nonpitting edema in one or more limbs. Diagnosis is by physical examination. Treatment consists of exercise, pressure gradient dressings, massage, and sometimes surgery. Cure is unusual, but treatment may lessen symptoms, slow progression, and prevent complications. Patients are at risk of cellulitis, lymphangitis, and, rarely, lymphangiosarcoma.
Etiology of Lymphedema
Lymphedema may be:
Primary: Due to lymphatic hypoplasia
Secondary: Due to obstruction or disruption of lymphatic vessels

Primary lymphedemas
Primary lymphedemas are often inherited and account for a small proportion of cases of lymphedema (1). They vary in phenotype and patient age at presentation but mostly involve the lower extremities (2). Classification is based on age at onset, although alternative phenotype-based classifications exist (3).
Congenital lymphedema appears before age 2 years and is due to lymphatic aplasia or hypoplasia. Milroy disease is an autosomal dominant familial form of congenital lymphedema, typically presenting with bilateral lower extremity edema, attributed to vascular endothelial growth factor receptor-3 (VEGFR-3) gene mutations (4). Other manifestations of Milroy disease include large veins in the lower leg, dysplastic toenails, and hydroceles in males. Intestinal lymphangiectasia with a protein-losing enteropathy, diarrhea, and cholestasis is less common.
Lymphedema praecox appears between ages 2 and 35 years, typically in females at the onset of menses or pregnancy (5). The term Meige disease, originally used to describe a form of lymphedema praecox accompanied by yellow nails, is now used as a more general term for lymphedema praecox (1, 6). Lymphedema praecox is usually autosomal dominant and involves painless swelling of one or both legs. Other findings can include extra eyelashes (distichiasis), cleft palate, and edema of legs, arms, and sometimes the face (3).
Lymphedema tarda occurs after age 35 years. Familial and sporadic forms exist; the genetic basis of both is unknown. Clinical findings are similar to those of lymphedema praecox but may be less severe.
Lymphedema is prominent in some other genetic syndromes, including:
Yellow nail syndrome, characterized by pleural effusions and yellow nails
Hennekam syndrome, a rare congenital syndrome of intestinal and other lymphangiectasia, facial anomalies, and intellectual disability (7)
Secondary lymphedema
Secondary lymphedema accounts for the vast majority of lymphedema cases (1, 8).
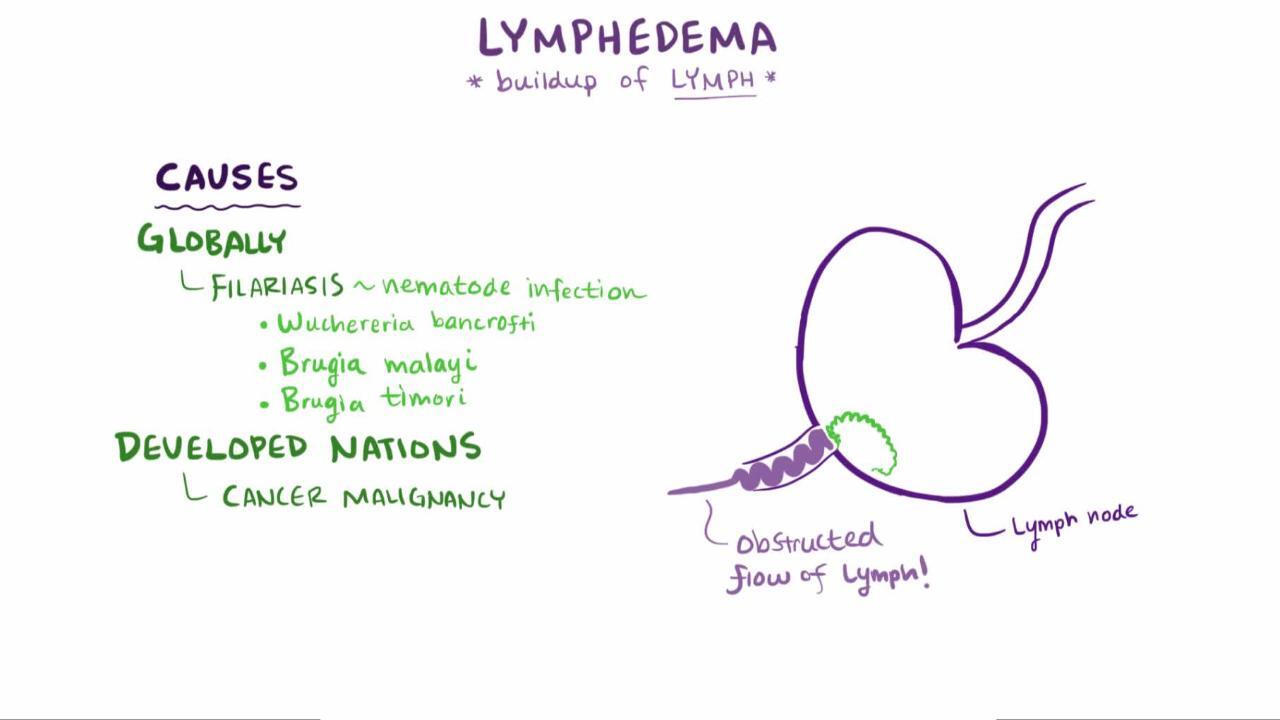
In the United States, the most common cause of secondary lymphedema is treatment for cancer (either surgical lymph node dissection or radiation therapy). In some of the world, particularly tropical and subtropical areas, however, lymphatic filariasis is the most common cause (9). Chronic venous insufficiency is a risk factor for lymphedema, due to leakage of lymph into interstitial tissues (9). Other important causes include direct vascular trauma and lymphatic obstruction by a tumor.
Etiology references
1. Brouillard P, Witte MH, Erickson RP, et al. Primary lymphoedema. Nat Rev Dis Primers. 2021;7(1):77. doi:10.1038/s41572-021-00309-7
2. Schook CC, Mulliken JB, Fishman SJ, Grant FD, Zurakowski D, Greene AK. Primary lymphedema: clinical features and management in 138 pediatric patients. Plast Reconstr Surg. 2011;127(6):2419-2431. doi:10.1097/PRS.0b013e318213a218
3. Connell F, Brice G, Jeffery S, Keeley V, Mortimer P, Mansour S. A new classification system for primary lymphatic dysplasias based on phenotype. Clin Genet. 2010;77(5):438-452. doi:10.1111/j.1399-0004.2010.01394.x
4. Van Zanten M, Mansour S, Ostergaard P, et al. Milroy Disease. 2006 Apr 27 [Updated 2021 Feb 18]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1239/
5. Shah AA, Petrosyan M, Nizam W, Roberson J, Guzzetta P. Resource overutilization in the diagnosis of lymphedema praecox. J Pediatr Surg. 2020;55(7):1363-1365. doi:10.1016/j.jpedsurg.2019.09.014
6. MedlinePlus [Internet]. Bethesda (MD): National Library of Medicine (US); Meige disease; [updated July 1, 2015]. Available from: https://medlineplus.gov/genetics/condition/noonan-syndrome/. Accessed January 12, 2026.
7. MedlinePlus [Internet]. Bethesda (MD): National Library of Medicine (US); Hennekam syndrome; [updated July 1, 2024]. Available from: Hennekam syndrome: MedlinePlus Genetics. Accessed January 27, 2026.
8. Rockson SG. Diagnosis and management of lymphatic vascular disease. J Am Coll Cardiol. 2008;52(10):799-806. doi:10.1016/j.jacc.2008.06.005
9. Lurie F, Malgor RD, Carman T, et al. The American Venous Forum, American Vein and Lymphatic Society and the Society for Vascular Medicine expert opinion consensus on lymphedema diagnosis and treatment. Phlebology. 2022;37(4):252-266. doi:10.1177/02683555211053532
Symptoms and Signs of Lymphedema
Symptoms of lymphedema include aching discomfort and a sensation of heaviness or fullness.
The cardinal sign is soft-tissue edema, graded in 3 progressive stages:
Stage 1: The edema is pitting, and the affected area often returns to normal by morning.
Stage 2: The edema is nonpitting, and chronic soft-tissue inflammation causes early fibrosis.
Stage 3: The edema is brawny and irreversible, largely because of soft-tissue fibrosis. In addition to being thickened, skin may have the dimpled appearance of an orange peel.
The swelling is most often unilateral and may worsen when the weather is warm, before menstruation occurs, and after the limb remains for a long time in a dependent position. It can affect any part of the limb (isolated proximal or distal) or the entire extremity; it can restrict range of motion when swelling is periarticular. Disability and emotional distress can be significant, especially when lymphedema results from medical or surgical treatment.

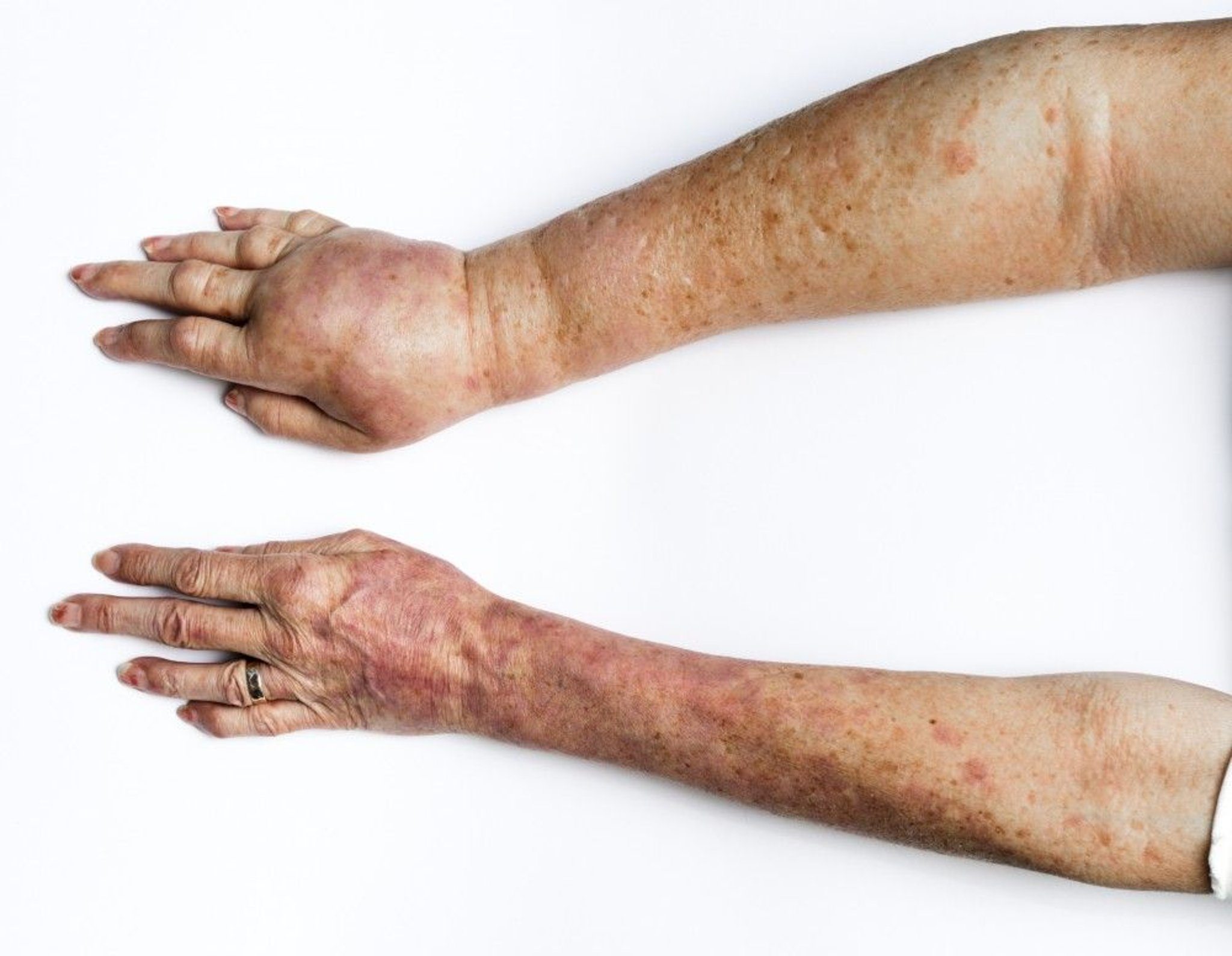
This photo shows the arms of a patient with lymphedema of the right (top) arm. Here, it is a result of surgery for breast cancer.
SCIENCE PHOTO LIBRARY
Other skin changes are common and include hyperkeratosis, hyperpigmentation, verrucae, papillomas, and fungal infections.

© Springer Science+Business Media
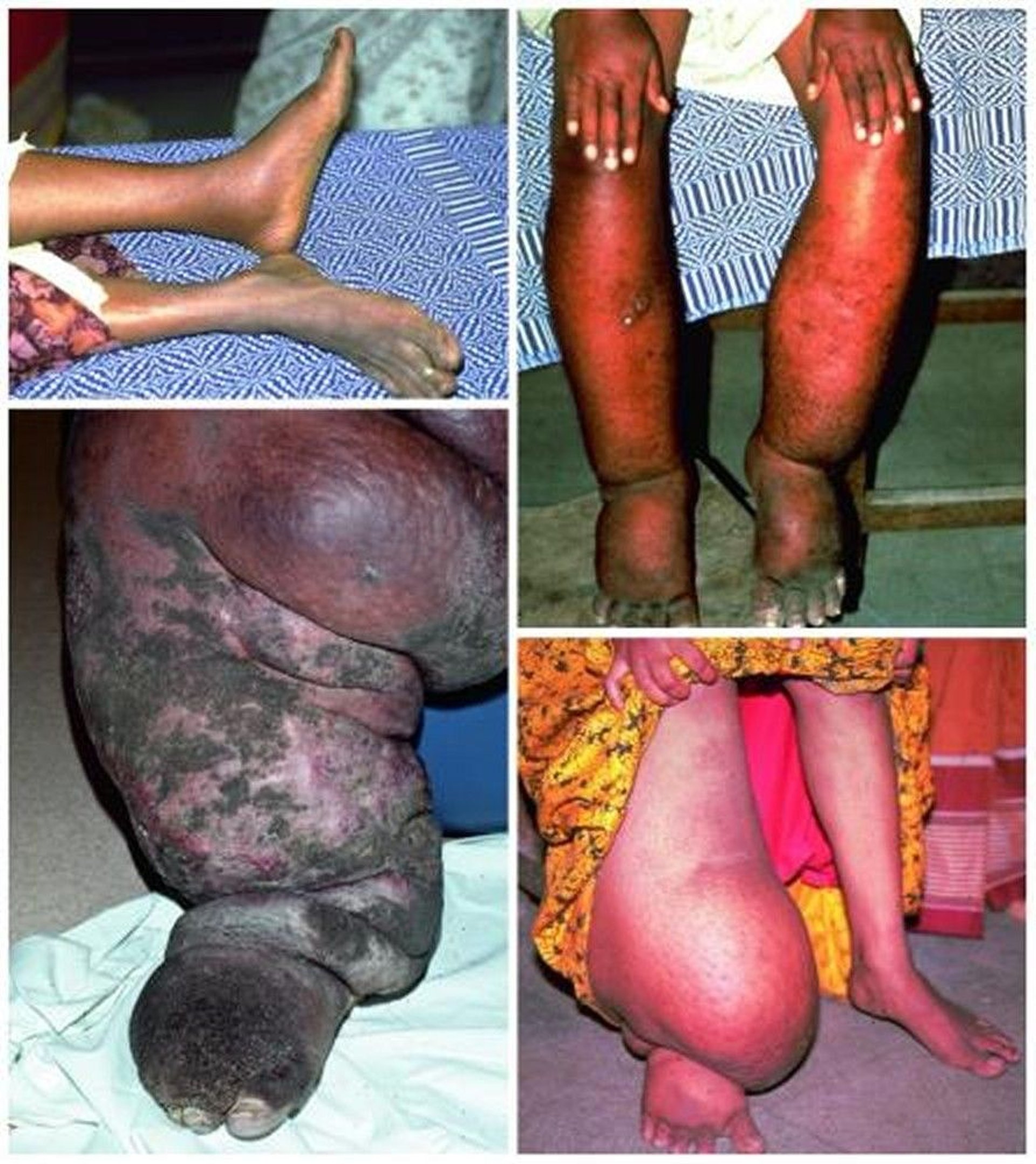
Rarely, an affected limb, or the scrotum, becomes extremely large, and the hyperkeratosis is severe, giving the appearance of elephant skin (elephantiasis). This manifestation is more common with filariasis than with other causes of lymphedema, and is related to repeated secondary bacterial infections (1, 2).
Complications
Lymphangitis may develop, most often when bacteria enter through skin cracks between the toes as a result of fungal infections or through cuts to the hand. Lymphangitis is usually streptococcal, causing erysipelas; sometimes it is staphylococcal. The affected limb becomes erythematous (possibly only dark on patients with dark skin) and feels hot; erythematous streaks may extend proximally from the point of entry, and lymphadenopathy may develop. Infrequently, the skin breaks down, which may lead to local infection or cellulitis.
Rarely, long-standing lymphedema leads to lymphangiosarcoma (Stewart-Treves syndrome), usually in patients who have had a mastectomy (3) or other cancer surgeries and radiation, and occasionally in patients with filariasis.
Symptoms and signs references
1. Centers for Disease Control and Prevention. Parasites. Filarial worms. Clinical Overview of Lymphatic Filariasis. My 13, 2024. Accessed January 12, 2026.
2. Taylor MJ, Hoerauf A, Bockarie M. Lymphatic filariasis and onchocerciasis. Lancet. 2010;376(9747):1175-1185. doi:10.1016/S0140-6736(10)60586-7
3. Kim PJ, Mufti A, Sachdeva M, et al. Stewart-Treves syndrome and other cutaneous malignancies in the context of chronic lymphedema: a systematic review. Int J Dermatol. 2022;61(1):62-70. doi:10.1111/ijd.15736
Diagnosis of Lymphedema
History and physical examination
Ultrasound
Sometimes lymphoscintigraphy, CT, or MRI
Diagnosis of primary lymphedema is usually based on characteristic soft-tissue edema throughout the body and other information from the history, including family history, and physical examination. Genetic testing may confirm the disease in the presence of family history with a known causative mutation, or be used to identify causative mutations in patients with suspected primary or syndromic lymphedema (1). Ultrasound is helpful to exclude other causes of lower extremity edema such as deep vein thrombosis (1).
Diagnosis of secondary lymphedema is usually obvious from physical examination. Ultrasound can be used to exclude deep vein thrombosis or other causes of lower extremity edema (2).
If the diagnosis or cause is not clear, radionuclide lymphoscintigraphy can identify lymphatic hypoplasia with sluggish flow of lymph, lymphatic aplasia, abnormal uptake into lymph nodes, and evidence of lymphatic hypertension (3). CT and MRI can evaluate secondary structural changes, and MR lymphography can show some lymphatic anomalies directly, either with or without contrast (4). CT and MRI can identify sites of lymphatic obstruction.
Tests for lymphatic filariasis should be performed in tropical and subtropical areas where the infecting microfilariae are present.
If lymphedema seems much greater than expected (eg, on the basis of lymph node dissection) or appears after a delay in a female treated for breast cancer, cancer recurrence should be considered.
Diagnosis references
1. Shah AA, Petrosyan M, Nizam W, Roberson J, Guzzetta P. Resource overutilization in the diagnosis of lymphedema praecox. J Pediatr Surg. 2020;55(7):1363-1365. doi:10.1016/j.jpedsurg.2019.09.014
2. Lurie F, Malgor RD, Carman T, et al. The American Venous Forum, American Vein and Lymphatic Society and the Society for Vascular Medicine expert opinion consensus on lymphedema diagnosis and treatment. Phlebology. 2022;37(4):252-266. doi:10.1177/02683555211053532
3. Rockson SG. Diagnosis and management of lymphatic vascular disease. J Am Coll Cardiol. 2008;52(10):799-806. doi:10.1016/j.jacc.2008.06.005
4. Executive Committee of the International Society of Lymphology. The diagnosis and treatment of peripheral lymphedema: 2020 Consensus Document of the International Society of Lymphology. Lymphology. 2020;53(1):3-19.
Treatment of Lymphedema
Manual lymphatic drainage, in which the limb is elevated above level of heart and compressed (“milked”) toward the heart
Limb exercises
External gradated compression stockings,, and intermittent pneumatic compression devices
Sometimes surgical soft-tissue reduction and reconstruction for primary lymphedema
Collectively, the combination of manual lymphatic drainage, external compression devices, exercises, and skin care is referred to as complex (or complete) decongestive therapy. This combination modality is recommended by experts and has been shown to be effective in reducing limb volume of the affected limb(s) (1, 2, 3).
Diuretic therapy is not indicated for lymphedema except in patients with comorbid conditions such (4). Dietary intervention is generally not helpful for lymphedema of the extremities; for chylous effusions or ascites caused by visceral lymphatic injury or lymphedema, a long-chain triglyceride-restricted diet may be helpful (3).
Cellulitis and lymphangitis are treated with beta-lactamase–resistant antibiotics that are effective against gram-positive organisms (eg, dicloxacillin).Cellulitis and lymphangitis are treated with beta-lactamase–resistant antibiotics that are effective against gram-positive organisms (eg, dicloxacillin).
In addition to the above treatments for lymphedema in general, treatment of secondary lymphedema involves managing its cause. Filariasis is treated with antiparasitic mediations (diethylcarbamazine, albendazole, ivermectin) (In addition to the above treatments for lymphedema in general, treatment of secondary lymphedema involves managing its cause. Filariasis is treated with antiparasitic mediations (diethylcarbamazine, albendazole, ivermectin) (5).
Surgical and microsurgical techniques such as liposuction, lymphatic reanastomoses, lymph node autotransplantation, formation of drainage channels, as well as surgical debulking procedures, are offered for patients who do not respond to more conservative measures but have not been rigorously studied (4).
Progression of disease can be monitored by measuring limb circumference, measuring water volume displaced by the submerged limb, or using skin or soft-tissue tonometry (to measure resistance to compression)
For patients who have undergone breast cancer surgery, surveillance, including serial bilateral arm circumference measurements or other measures are recommended (6).
Secondary preventive measures include avoiding application of heat, vigorous exercise, being sedentary for long periods, and constrictive garments (including blood pressure cuffs) around the affected limb (7). Skin and nail care require meticulous attention; vaccination, phlebotomy, and IV catheterization in an affected limb should be avoided.
Multiple pharmacologic options are under study for both primary and secondary lymphedema (1, 4). These include antiproliferative medications for infants and young children with syndromic lymphedema. Pharmacologic therapy should be individualized and considered in consultation with a vascular or lymphatic specialist.
Pearls & Pitfalls
|
Treatment references
1. Brouillard P, Witte MH, Erickson RP, et al. Primary lymphoedema. Nat Rev Dis Primers. 2021;7(1):77. doi:10.1038/s41572-021-00309-7
2. Lurie F, Malgor RD, Carman T, et al. The American Venous Forum, American Vein and Lymphatic Society and the Society for Vascular Medicine expert opinion consensus on lymphedema diagnosis and treatment. Phlebology. 2022;37(4):252-266. doi:10.1177/02683555211053532
3. Rockson SG. Diagnosis and management of lymphatic vascular disease. J Am Coll Cardiol. 2008;52(10):799-806. doi:10.1016/j.jacc.2008.06.005
4. Executive Committee of the International Society of Lymphology. The diagnosis and treatment of peripheral lymphedema: 2020 Consensus Document of the International Society of Lymphology. Lymphology. 2020;53(1):3-19.
5. Shah AA, Petrosyan M, Nizam W, Roberson J, Guzzetta P. Resource overutilization in the diagnosis of lymphedema praecox. J Pediatr Surg. 2020;55(7):1363-1365. doi:10.1016/j.jpedsurg.2019.09.014
6. McLaughlin SA, Staley AC, Vicini F, et al. Considerations for Clinicians in the Diagnosis, Prevention, and Treatment of Breast Cancer-Related Lymphedema: Recommendations from a Multidisciplinary Expert ASBrS Panel : Part 1: Definitions, Assessments, Education, and Future Directions. Ann Surg Oncol. 2017;24(10):2818-2826. doi:10.1245/s10434-017-5982-4
7. Asdourian MS, Skolny MN, Brunelle C, Seward CE, Salama L, Taghian AG. Precautions for breast cancer-related lymphoedema: risk from air travel, ipsilateral arm blood pressure measurements, skin puncture, extreme temperatures, and cellulitis. Lancet Oncol. 2016;17(9):e392-e405. doi:10.1016/S1470-2045(16)30204-2
Prognosis for Lymphedema
Cure is unusual once lymphedema occurs (1, 2). Meticulous treatment and possibly preventive measures can lessen symptoms, slow or halt disease progression, and prevent complications.
Prognosis references
1. Brouillard P, Witte MH, Erickson RP, et al. Primary lymphoedema. Nat Rev Dis Primers. 2021;7(1):77. doi:10.1038/s41572-021-00309-7
2. Rockson SG. Diagnosis and management of lymphatic vascular disease. J Am Coll Cardiol. 2008;52(10):799-806. doi:10.1016/j.jacc.2008.06.005
Key Points
Secondary lymphedema (due to obstruction or disruption of lymphatic vessels) is far more common than primary lymphedema (which is due to lymphatic aplasia/hypoplasia).
Elephantiasis (extreme hyperkeratosis of skin in a limb affected by lymphedema) is a severe manifestation of lymphedema usually due to lymphatic filariasis.
Cure of lymphedema is unusual, but complex decongestive therapy can decrease symptoms, slow or halt disease progression, and prevent complications.
More Information
The following English-language resources may be useful. Please note that The Manual is not responsible for the content of these resources.
National Lymphedema Network. Position papers intended to standardize clinical education and practice
McLaughlin SA, Staley AC, Vicini F, et al. Considerations for Clinicians in the Diagnosis, Prevention, and Treatment of Breast Cancer-Related Lymphedema: Recommendations from a Multidisciplinary Expert ASBrS Panel : Part 1: Definitions, Assessments, Education, and Future Directions. Ann Surg Oncol 2017;24(10):2818-2826. doi:10.1245/s10434-017-5982-4
Drug Information for the Topic





