

Mastocytosis is mast cell proliferation with infiltration of skin or other tissues and organs. Mast cell activation syndrome is increased and inappropriate activation of mast cells without clonal proliferation. Symptoms result mainly from mediator release and include pruritus, flushing, and dyspepsia due to gastric hypersecretion. Diagnosis is by skin or bone marrow biopsy or both. Treatment is with antihistamines and control of any underlying disorder.
(See also Overview of Allergic and Atopic Disorders.)
Mastocytosis is a group of disorders characterized by proliferation of mast cells and infiltration of the skin, other organs, or both. Pathology results mainly from release of mast cell mediators, including histamine, heparin, leukotrienes, and various inflammatory cytokines. Histamine causes many symptoms, including gastric symptoms, but other mediators also contribute. Significant organ infiltration may cause organ dysfunction. Mediator release may be triggered by physical touch, exercise, alcohol, nonsteroidal anti-inflammatory drugs (NSAIDs), opioids, insect stings, or foods.
Etiology in many cases of mastocytosis involves an activating mutation (D816V) in the gene coding for the stem cell factor receptor c-kit, which is present on mast cells. The result is autophosphorylation of the receptor, which causes uncontrolled mast cell proliferation.
Classification of Mastocytosis
Mastocytosis may be cutaneous or systemic.
Cutaneous mastocytosis
Cutaneous mastocytosis typically occurs in children. Most patients present with urticaria pigmentosa, a local or diffusely distributed salmon or brown maculopapular rash caused by multiple small mast cell collections. Nodular lesions and plaques can also develop. Less common are diffuse cutaneous mastocytosis, which is skin infiltration without discrete lesions, and mastocytoma, which is a large (1 to 5 cm) solitary collection of mast cells.
Cutaneous forms rarely progress to systemic disease in children but may do so in adults.

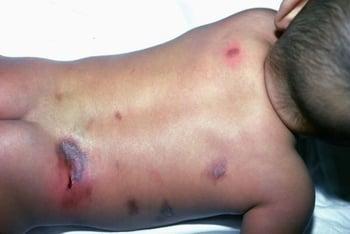
This image shows characteristic mastocytomas.

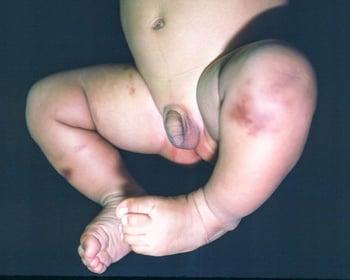
This image shows the plaque-like appearance of cutaneous mastocytosis.

Lesions consist of reddish brown, irregular plaques that urticate when rubbed (Darier sign).
Lesions consist of reddish brown, irregular plaques that urticate when rubbed (Darier sign).
By permission of the publisher. From Joe E, Soter N. In Current Dermatologic Diagnosis and Treatment, edited by I Freedberg, IM Freedberg, and MR Sanchez. Philadelphia, Current Medicine, 2001.

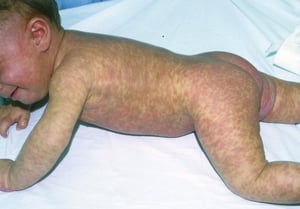
The infant shown here has profuse papulonodular and plaque lesions of urticaria pigmentosa.
The infant shown here has profuse papulonodular and plaque lesions of urticaria pigmentosa.
© Springer Science+Business Media

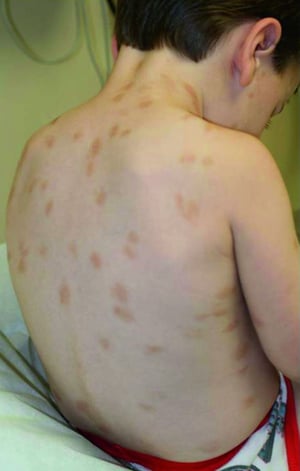
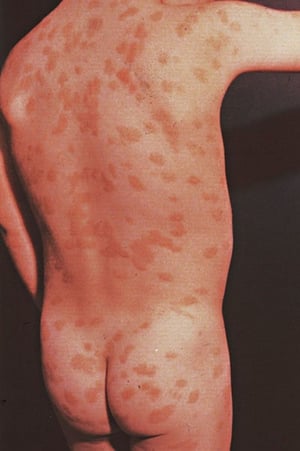
This photo shows reddish brown macules on the back of a school-aged child.
This photo shows reddish brown macules on the back of a school-aged child.
© Springer Science+Business Media

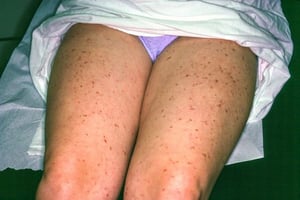
This image shows yellowish tan to reddish brown macules and papules of urticaria pigmentosa.
This image shows yellowish tan to reddish brown macules and papules of urticaria pigmentosa.
Image courtesy of Karen McKoy, MD.

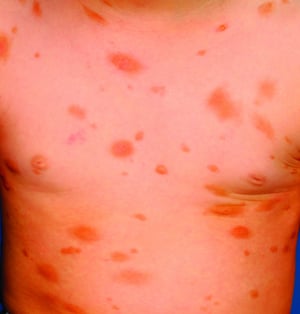
Urticaria pigmentosa may manifest as erythematous plaque-like lesions on the skin.
Urticaria pigmentosa may manifest as erythematous plaque-like lesions on the skin.
© Springer Science+Business Media
Lesions consist of reddish brown, irregular plaques that urticate when rubbed (Darier sign).
Lesions consist of reddish brown, irregular plaques that urticate when rubbed (Darier sign).
By permission of the publisher. From Joe E, Soter N. In Current Dermatologic Diagnosis and Treatment, edited by I Freedberg, IM Freedberg, and MR Sanchez. Philadelphia, Current Medicine, 2001.
The infant shown here has profuse papulonodular and plaque lesions of urticaria pigmentosa.
The infant shown here has profuse papulonodular and plaque lesions of urticaria pigmentosa.
© Springer Science+Business Media
This photo shows reddish brown macules on the back of a school-aged child.
This photo shows reddish brown macules on the back of a school-aged child.
© Springer Science+Business Media
This image shows yellowish tan to reddish brown macules and papules of urticaria pigmentosa.
This image shows yellowish tan to reddish brown macules and papules of urticaria pigmentosa.
Image courtesy of Karen McKoy, MD.
Urticaria pigmentosa may manifest as erythematous plaque-like lesions on the skin.
Urticaria pigmentosa may manifest as erythematous plaque-like lesions on the skin.
© Springer Science+Business Media
Systemic mastocytosis
Systemic mastocytosis most commonly occurs in adults and is characterized by multifocal bone marrow lesions; it often involves other organs, most commonly skin, lymph nodes, liver, spleen, and/or gastrointestinal (GI) tract.
Systemic mastocytosis is classified as
Indolent mastocytosis, with no organ dysfunction and a good prognosis
Mastocytosis associated with other hematologic disorders (eg, myeloproliferative disorders, myelodysplasia, lymphoma)
Aggressive mastocytosis, characterized by impaired organ function
Mast cell leukemia, with > 20% mast cells in bone marrow, no skin lesions, multiorgan failure, and a poor prognosis
Mast cell activation syndrome
Mast cell activation syndrome is characterized by increased and inappropriate activation of mast cells with mediator release but without clonal proliferation or organ infiltration by mast cells (1). The syndrome was originally diagnosed only when mediator release was idiopathic, but it has since been expanded to include release triggered by allergen-specific IgE, certain medications, or physical factors. Genetic causes are suspected but not proved. Most cases do not involve clonal proliferation of mast cells but are due to a lower threshold for mast cells to degranulate. Mast cell activation syndrome has frequently been associated with postural orthostatic tachycardia syndrome (POTS) and Ehlers-Danlos syndrome, although the nature of the connection is unclear.
The manifestations of mast cell activation syndrome are frequently similar to those of patients with systemic mastocytosis; they include tachycardia, syncope, urticaria, flushing, nausea, vomiting, and brain fog.
It is unclear whether mast cell activation syndrome can progress to systemic mastocytosis or another form of mast cell disease and, if so, how many patients are affected.
Classification reference
1. Weiler CR, Austen KF, Akin C, et al: AAAAI Mast Cell Disorders Committee Work Group Report: Mast cell activation syndrome (MCAS) diagnosis and management. J Allergy Clin Immunol 144 (4):883–896, 2019. doi: 10.1016/j.jaci.2019.08.023
Symptoms and Signs of Mastocytosis
Skin involvement is often pruritic in mastocytosis—whether a single mastocytoma or more diffuse disease. The following may worsen itching:
Changes in temperature
Contact with clothing or other materials
Use of some medications, including nonsteroidal anti-inflammatory drugs (NSAIDs)
Consumption of hot beverages, spicy foods, or alcohol
Exercising
Stroking or rubbing skin lesions causes urticaria and erythema around the lesion (Darier sign); this reaction differs from dermatographism, which involves normal skin.

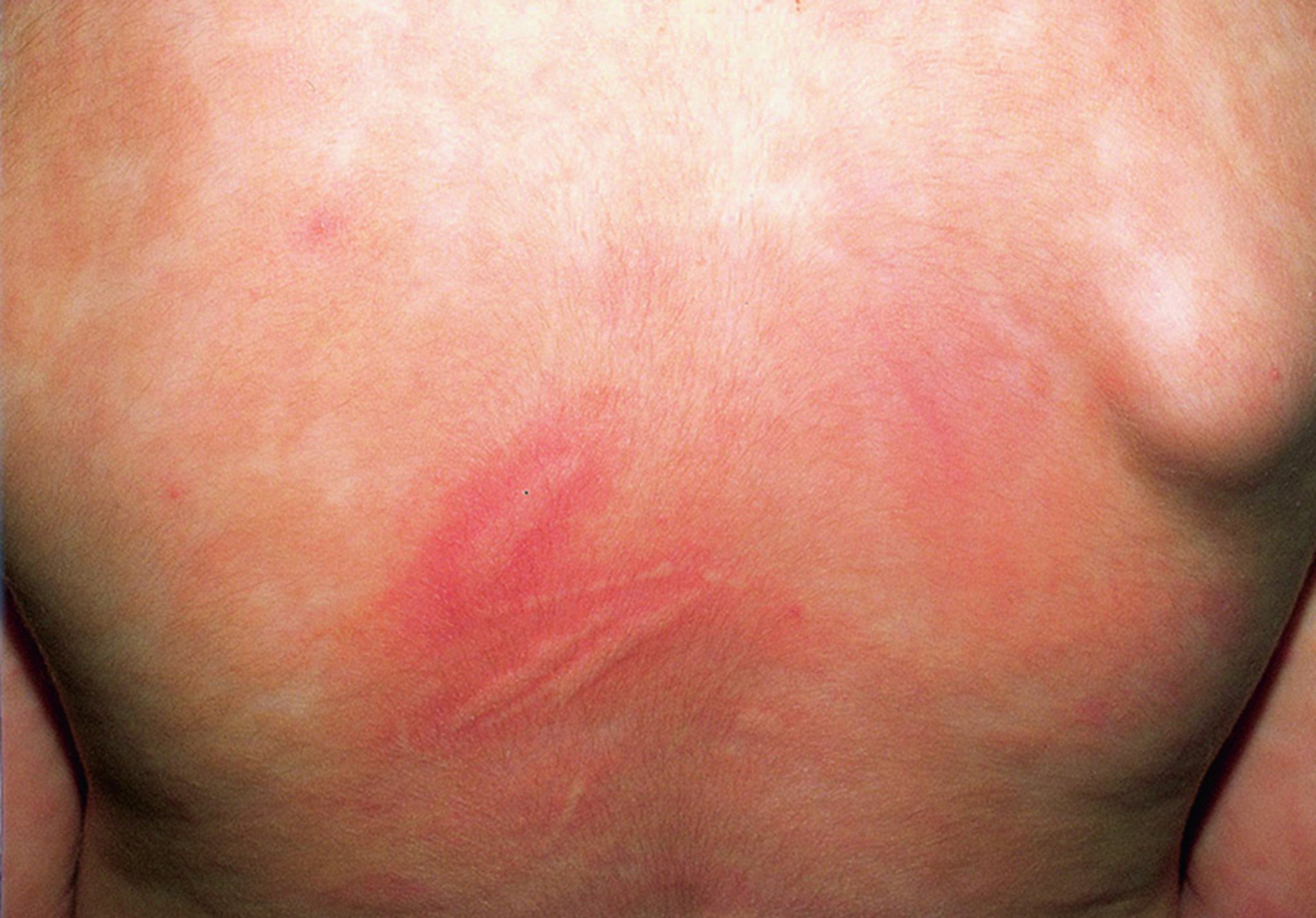
This photo shows the characteristic finding of transient urticarial swelling around the lesion site, known as Darier sign.
© Springer Science+Business Media
Systemic symptoms can occur with any form. The most common is flushing; the most dramatic are anaphylactoid and anaphylactic reactions with syncope and shock.
Other symptoms include epigastric pain due to peptic ulcer disease, nausea (because histamine stimulates gastric acid production), vomiting, chronic diarrhea, arthralgias, bone pain, and neuropsychiatric changes (eg, irritability, depression, mood lability). Hepatic and splenic infiltration may cause portal hypertension with resultant ascites.
Diagnosis of Mastocytosis
History and physical examination
Bone marrow biopsy
Serum tryptase levels (baseline and during symptoms if possible)
Systemic mastocytosis
Diagnosis of mastocytosis is suggested by clinical presentation. However, similar symptoms can be caused by many other disorders such as carcinoid syndrome, vipoma, gastrinoma (Zollinger-Ellison syndrome), and chronic urticaria.
Diagnosis requires bone marrow biopsy and tryptase levels (a marker of mast cell degranulation) in most patients. Skin biopsy can be done to check for mast cells, but this test does not replace the need for a bone marrow biopsy to classify the diagnosis and staging.
Diagnosis of mastocytosis is confirmed when one major criterion and at least one minor criterion or ≥ 3 (of 4) minor criteria are met.
The major criterion is
The presence of multifocal, dense aggregates of > 15 mast cells in bone marrow (preferred) or other extracutaneous organs (except the GI tract, lymph nodes, liver, or spleen)
The 4 minor criteria are
Atypical morphology or spindle shapes in > 25% of the mast cells in bone marrow biopsy sections or aspirate
A kit mutation at codon 816 (commonly Asp816Val) in bone marrow, peripheral blood, or other tissue
Bone marrow or other extracutaneous mast cells expressing the surface markers CD2, CD25, or both
Baseline serum tryptase levels > 20 ng/mL (> 20 mcg/L); values > 11.4 ng/mL (11.4 mcg/L) are considered elevated in most diagnostic laboratories
The baseline level of tryptase is elevated in systemic mastocytosis but is typically normal in cutaneous mastocytosis and mast cell activation syndrome.
Mast cell activation syndrome
The diagnosis of mast cell activation syndrome requires meeting all 3 criteria (1):
Symptoms consistent with mast cell degranulation and release of mast cell mediators
Laboratory evidence of an increase in mast cell mediators in the serum or urine such as an increase in serum tryptase during an event of 20% + 2 ng/mL above baseline or an increase in urinary metabolites such as N-methylhistamine, prostaglandin D2, or leukotriene E4 (2)
Positive clinical response to anti-mast cell therapy
Patients with mast cell activation typically have a normal bone marrow biopsy if it is done.
If the diagnosis is uncertain, levels of mast cell mediators and their metabolites (eg, 24-hour N-methylhistamine, prostaglandin D2, leukotriene E4) may be measured in plasma and urine; elevated levels support the diagnosis of mast cell disease but not necessarily systemic mastocytosis.
A bone scan and GI evaluation can also be helpful in cases where the diagnosis requires confirmation.
Diagnosis references
1. Valent P, Akin C, Hartmann K, et al: Updated Diagnostic Criteria and Classification of Mast Cell Disorders: A Consensus Proposal. Hemasphere 5(11):e646, 2021. doi:10.1097/HS9.0000000000000646
2. Leru PM, Anton VF, Ureche C, Zurac S, Bratu O, Neagoe CD: Mast cell activation syndromes - evaluation of current diagnostic criteria and laboratory tools in clinical practice (Review). Exp Ther Med 20(3):2348–2351, 2020. doi:10.3892/etm.2020.8947
Treatment of Mastocytosis
For cutaneous mastocytosis, H1 blockers and possibly psoralen plus ultraviolet light or topical corticosteroids
For systemic mastocytosis and mast cell activation syndrome, H1 and H2 blockers, cromolyn, ketotifen, montelukast, and aspirinFor systemic mastocytosis and mast cell activation syndrome, H1 and H2 blockers, cromolyn, ketotifen, montelukast, and aspirin
For aggressive forms, midostaurin, avapritinib, interferon alfa-2b, or corticosteroidsFor aggressive forms, midostaurin, avapritinib, interferon alfa-2b, or corticosteroids
Cutaneous mastocytosis
H1 blockers are effective for symptoms. Children with cutaneous forms require no additional treatment because most cases resolve spontaneously.
Adults with cutaneous forms may be treated with psoralen plus ultraviolet light or with topical corticosteroids once or twice a day.
Mastocytoma usually involutes spontaneously and requires no treatment.
Systemic mastocytosis
Management of anaphylactic reactions includes parenteral epinephrine, inhaled beta-agonists for wheezing, and IV fluid replacement for hypotension. includes parenteral epinephrine, inhaled beta-agonists for wheezing, and IV fluid replacement for hypotension.
All patients with systemic mastocytosis should be treated with H1 and H2 blockers and should carry a prefilled, self-injecting epinephrine syringe. All patients with systemic mastocytosis should be treated with H1 and H2 blockers and should carry a prefilled, self-injecting epinephrine syringe.
Aspirin controls flushing but may enhance leukotriene production, thereby contributing to other mast cell–related symptoms; it should not be given to children because Reye syndrome is a risk.Aspirin controls flushing but may enhance leukotriene production, thereby contributing to other mast cell–related symptoms; it should not be given to children because Reye syndrome is a risk.
Cromolyn may help by preventing mast cell degranulation. Ketotifen may also be effective. No treatment can reduce the number of tissue mast cells.Cromolyn may help by preventing mast cell degranulation. Ketotifen may also be effective. No treatment can reduce the number of tissue mast cells.
Omalizumab is an anti-IgE antibody that is sometimes used for moderate to severe asthma and chronic urticaria; it has sometimes been used in patients with mastocytosis or mast cell activation syndrome to try to prevent anaphylaxis.Omalizumab is an anti-IgE antibody that is sometimes used for moderate to severe asthma and chronic urticaria; it has sometimes been used in patients with mastocytosis or mast cell activation syndrome to try to prevent anaphylaxis.
In patients with an aggressive form of mastocytosis, characterized by increasing mast cell accumulation in different organs, leading to dysfunction, the multikinase inhibitors midostaurin or avapritinib (In patients with an aggressive form of mastocytosis, characterized by increasing mast cell accumulation in different organs, leading to dysfunction, the multikinase inhibitors midostaurin or avapritinib (1) can be used to help control end-organ damage, cytopenias, and mast cell accumulation in bone marrow (2).
Avapritinib can also be used for symptomatic management in indolent mastocytosis (Avapritinib can also be used for symptomatic management in indolent mastocytosis (3).
Interferon alfa-2b induces regression of bone lesions. Corticosteroids may be required as adjunctive treatment for severe cases. Interferon alfa-2b induces regression of bone lesions. Corticosteroids may be required as adjunctive treatment for severe cases.
Cytotoxic agents (eg, daunomycin, etoposide, 6-mercaptopurine) may be indicated for treatment of mast cell leukemia, but efficacy is unproved. Imatinib (a tyrosine kinase receptor inhibitor) may be useful when treating adults with aggressive systemic mastocytosis but is ineffective in patients with the D816V Cytotoxic agents (eg, daunomycin, etoposide, 6-mercaptopurine) may be indicated for treatment of mast cell leukemia, but efficacy is unproved. Imatinib (a tyrosine kinase receptor inhibitor) may be useful when treating adults with aggressive systemic mastocytosis but is ineffective in patients with the D816Vc-kit mutation.
Mast cell activation syndrome
Treatment is directed at preventing mediator release (eg, with cromolyn and/or ketotifen) and blocking mediator effects with some regimen of H1 and H2 blockers to block histamine, aspirin to block prostaglandins and montelukast to block leukotrienes. Treatment is directed at preventing mediator release (eg, with cromolyn and/or ketotifen) and blocking mediator effects with some regimen of H1 and H2 blockers to block histamine, aspirin to block prostaglandins and montelukast to block leukotrienes.
Known triggers should be avoided.
Treatment references
1. Radia D, Deininger M, Gotlib J, et al: Avapritinib, a potent and selective inhibitor of KIT D816V, induces complete and durable responses in patients (pts) with advanced systemic mastocytosis (AdvSM). EHA Library 2019;267413:S830.
2. Gotlib J, Kluin-Nelemans HC, George TI, et al: Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med 374 (26):2530–2541, 2016. doi: 10.1056/NEJMoa1513098
3. Gotlib J, Castells M, Elberink HO, et al: Avapritinib versus Placebo in Indolent Systemic Mastocytosis. NEJM Evid 2(6):EVIDoa2200339, 2023. doi:10.1056/EVIDoa2200339
Key Points
Patients with cutaneous mastocytosis, usually children, typically present with a diffuse salmon or brown, often pruritic maculopapular rash.
Systemic mastocytosis causes multifocal bone marrow lesions, usually in adults, but often affects other organs.
All types can cause systemic symptoms (most commonly, flushing but sometimes anaphylactoid reactions).
For cutaneous mastocytosis, use H1 blockers to relieve symptoms, and in adults, consider treatment with psoralen plus ultraviolet light or topical corticosteroids.
For systemic mastocytosis, use H1 and H2 blockers and sometimes cromolyn, and for aggressive mastocytosis, consider midostaurin, avapritinib, interferon alfa-2b, systemic corticosteroids.For systemic mastocytosis, use H1 and H2 blockers and sometimes cromolyn, and for aggressive mastocytosis, consider midostaurin, avapritinib, interferon alfa-2b, systemic corticosteroids.
For mast cell activation syndrome, target mediators released during activation with antihistamines, leukotriene inhibitors, and mast cell stabilizers.
Make sure all patients with mastocytosis carry a prefilled, self-injecting epinephrine syringe.Make sure all patients with mastocytosis carry a prefilled, self-injecting epinephrine syringe.
Drug Information for the Topic





