

Leprosy is a chronic infection caused by the acid-fast bacilli Mycobacterium leprae or the closely related organism M. lepromatosis. These organisms have a unique tropism for peripheral nerves, skin, and mucous membranes of the upper respiratory tract. Symptoms are myriad and include anesthetic polymorphic skin lesions and peripheral neuropathy. Diagnosis is primarily clinical and confirmed by molecular tests and biopsy. Treatment is typically with dapsone, rifampin (rifampicin), and clofazimine. Patients rapidly become noncontagious after starting therapy. . These organisms have a unique tropism for peripheral nerves, skin, and mucous membranes of the upper respiratory tract. Symptoms are myriad and include anesthetic polymorphic skin lesions and peripheral neuropathy. Diagnosis is primarily clinical and confirmed by molecular tests and biopsy. Treatment is typically with dapsone, rifampin (rifampicin), and clofazimine. Patients rapidly become noncontagious after starting therapy.
M. leprae was the only known cause of leprosy until 2008, when a second species, M. lepromatosis, was identified in Mexico. Together, these two organisms are called M. leprae complex.
In much of the world, leprosy is rare. It is a complex, poorly characterized disease, and its pathogenesis is challenging to study.
Although leprosy is not highly contagious (contrary to popular belief), rarely causes death, and can be effectively treated with antibiotics, it continues to be associated with considerable social stigma. Misunderstanding about the disease probably exists because leprosy was incurable before the advent of effective antibiotic therapy in the 1940s. Prior to therapy, people with the disease would become disfigured and often have significant disability, which caused others to fear and shun them. Because of this social stigma, the psychological impact of leprosy is often significant.
Epidemiology of Leprosy
In 2023, 182,815 new cases of leprosy were reported globally, and most cases occurred in India, Brazil, and Indonesia. Among all cases, 13.6% of patients were from the Americas, of which > 90% were from Brazil (1).
In 2024, 205 new cases were reported in the United States; almost two-thirds occurred in Florida, Louisiana, Massachusetts, Texas, Hawaii, California, New York, Georgia, and Illinois (2). Most cases of leprosy in the United States involve people who emigrated from or worked in countries where leprosy is common. Most of the indigenously acquired cases in the United States involved people who live in southern states where nine-banded armadillos infected with unique genotypes of M. leprae are found and who reported direct contact with armadillos (3).
Leprosy can develop at any age. Older age is a risk factor, but disease occurs most often in people aged 5 to 15 years or > 30 years.
Leprosy is not highly transmissible, and approximately 95% of people who are exposed to M. leprae never develop leprosy (4).
Epidemiology references
1. Pan American Health Organization (PAHO): Leprosy (Hansen disease). Accessed October 29, 2025.
2. Health Resources and Services Administration: National Hansen's Disease (Leprosy) Program. August 2025. Accessed September 17, 2025.
3. Truman RW, Singh P, Sharma R, et al. Probable zoonotic leprosy in the southern United States. N Engl J Med. 2011;364(17):1626–1633. doi:10.1056/NEJMoa1010536
4. MedlinePlus [Internet]. Bethesda (MD): National Library of Medicine (US); [updated Jun 24]. Leprosy; [updated 2018 Feb 1; reviewed 2020 Apr 1; cited 2025 Oct 3]; [about 5 p.]. Available from: Leprosy: MedlinePlus Genetic Conditions.
Pathophysiology of Leprosy
Humans are the main natural reservoir for M. leprae. Armadillos are one confirmed source of zoonotic transmission to humans, but there are reports of M. leprae in red squirrels in the British Isles (1). Natural leprosy infections have also been reported in nonhuman primates in the wild (eg, wild chimpanzees, sooty mangabeys, cynomolgus macaques), but it is unclear whether these lead to zoonotic transmission to humans. (See also Mycobacterial Infections in Animals.)
Leprosy is thought to be transmitted from person to person through nasal droplets and secretions. Transmission does not appear to occur through casual contact (eg, simply touching someone with the infection) or short-term contact. People who live in households where they are in contact with known infected people have an approximately 2-fold increased risk of leprosy (2).
Even after contact with the bacteria, most people do not develop leprosy (3); health care professionals often work for many years with people who have leprosy without contracting it. Most immunocompetent people who are infected with M. leprae do not develop leprosy. People who do develop leprosy may have a genetic predisposition, but this has not been well-defined.
M. leprae grow slowly (doubling in 2 weeks). The usual incubation period ranges from a few months to 20 years (3). Once infection develops, hematogenous dissemination can occur.
Classification of leprosy
Leprosy is classified by type and number of skin areas affected:
Paucibacillary: ≤ 5 skin lesions with no bacteria detected on samples from those areas
Multibacillary: ≥ 6 skin lesions, bacteria detected on samples from skin lesions, or both
Leprosy also is classified by cellular response and clinical findings:
Tuberculoid
Lepromatous
Borderline
People with tuberculoid leprosy typically have a strong cell-mediated response, which limits disease to a few skin lesions (paucibacillary), and the disease is milder, less common, and less contagious.
People with lepromatous or borderline leprosy typically have poor cell-mediated immunity to M. leprae and have more severe, systemic infection with widespread bacterial infiltration of skin, nerves, and other organs (eg, nose, testes, kidneys). They usually have more skin lesions (multibacillary), and the disease is more contagious.
In both classifications, the type of leprosy dictates:
Long-term prognosis
Likely complications
Duration of antibiotic treatment
Pathophysiology references
1. Ploemacher T, Faber WR, Menke H, Rutten V, Pieters T. Reservoirs and transmission routes of leprosy; A systematic review. PLoS Negl Trop Dis. 2020;14(4):e0008276. Published 2020 Apr 27. doi:10.1371/journal.pntd.0008276
2. Fine PE, Sterne JA, Pönnighaus JM, et al. Household and dwelling contact as risk factors for leprosy in northern Malawi. Am J Epidemiol. 1997;146(1):91-102. doi:10.1093/oxfordjournals.aje.a009195
3. MedlinePlus [Internet]. Bethesda (MD): National Library of Medicine (US); [updated Jun 24]. Leprosy; [updated 2018 Feb 1; reviewed 2020 Apr 1; cited 2025 Oct 3]; [about 5 p.]. Available from: Leprosy: MedlinePlus Genetic Conditions.
Symptoms and Signs of Leprosy
Symptoms of leprosy usually do not begin until > 1 year after infection. Once symptoms begin, they progress slowly.


This photo shows skin lesions consisting of hypopigmented macules with overlying numbness due to nerve damage.
This photo shows skin lesions consisting of hypopigmented macules with overlying numbness due to nerve damage.
CNRI/SCIENCE PHOTO LIBRARY


The photo shows skin lesions consisting of symmetric nodules and skin thickening. The eyebrows are missing.
The photo shows skin lesions consisting of symmetric nodules and skin thickening. The eyebrows are missing.
ST MARY'S HOSPITAL MEDICAL SCHOOL/SCIENCE PHOTO LIBRARY


This photo shows features of borderline leprosy that is predominantly tuberculoid in appearance.
This photo shows features of borderline leprosy that is predominantly tuberculoid in appearance.
Photo courtesy of Karen McKoy, MD.
This photo shows skin lesions consisting of hypopigmented macules with overlying numbness due to nerve damage.
This photo shows skin lesions consisting of hypopigmented macules with overlying numbness due to nerve damage.
CNRI/SCIENCE PHOTO LIBRARY
The photo shows skin lesions consisting of symmetric nodules and skin thickening. The eyebrows are missing.
The photo shows skin lesions consisting of symmetric nodules and skin thickening. The eyebrows are missing.
ST MARY'S HOSPITAL MEDICAL SCHOOL/SCIENCE PHOTO LIBRARY
This photo shows features of borderline leprosy that is predominantly tuberculoid in appearance.
This photo shows features of borderline leprosy that is predominantly tuberculoid in appearance.
Photo courtesy of Karen McKoy, MD.
Leprosy affects mainly the skin and peripheral nerves. Nerve involvement causes numbness and weakness in areas controlled by the affected nerves.
Tuberculoid leprosy: Skin lesions consist of one or a few hypoesthetic, centrally hypopigmented macules with sharp, raised borders. The rash, as in all forms of leprosy, is nonpruritic. Areas affected by this rash are numb because of damage to the underlying peripheral nerves, which may be palpably enlarged.
Lepromatous leprosy: Much of the skin and many areas of the body, including the kidneys, nose, and testes, may be affected. Patients have skin macules, papules, nodules, or plaques, which are often symmetric. Peripheral neuropathy is more severe than in tuberculoid leprosy, with more areas of numbness; certain muscle groups may be weak. Patients may develop gynecomastia or lose eyelashes and eyebrows.
Borderline leprosy: Features of both tuberculoid and lepromatous leprosy are present. Without treatment, borderline leprosy may become less severe and more like the tuberculoid form, or it may worsen and become more like the lepromatous form.
Complications of leprosy
The most severe complications result from the peripheral neuropathy, which causes deterioration of the sense of touch and a corresponding inability to feel pain and temperature. Patients may unknowingly burn, cut, or otherwise harm themselves. Repeated damage may lead to loss of digits. Muscle weakness can result in deformities (eg, clawing of the fourth and fifth digits caused by ulnar nerve involvement, foot drop caused by peroneal nerve involvement).

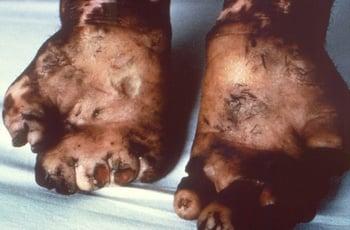
This photo shows severe mutilation and degeneration of digits in a patient with leprosy.
Papules and nodules can be particularly disfiguring on the face.

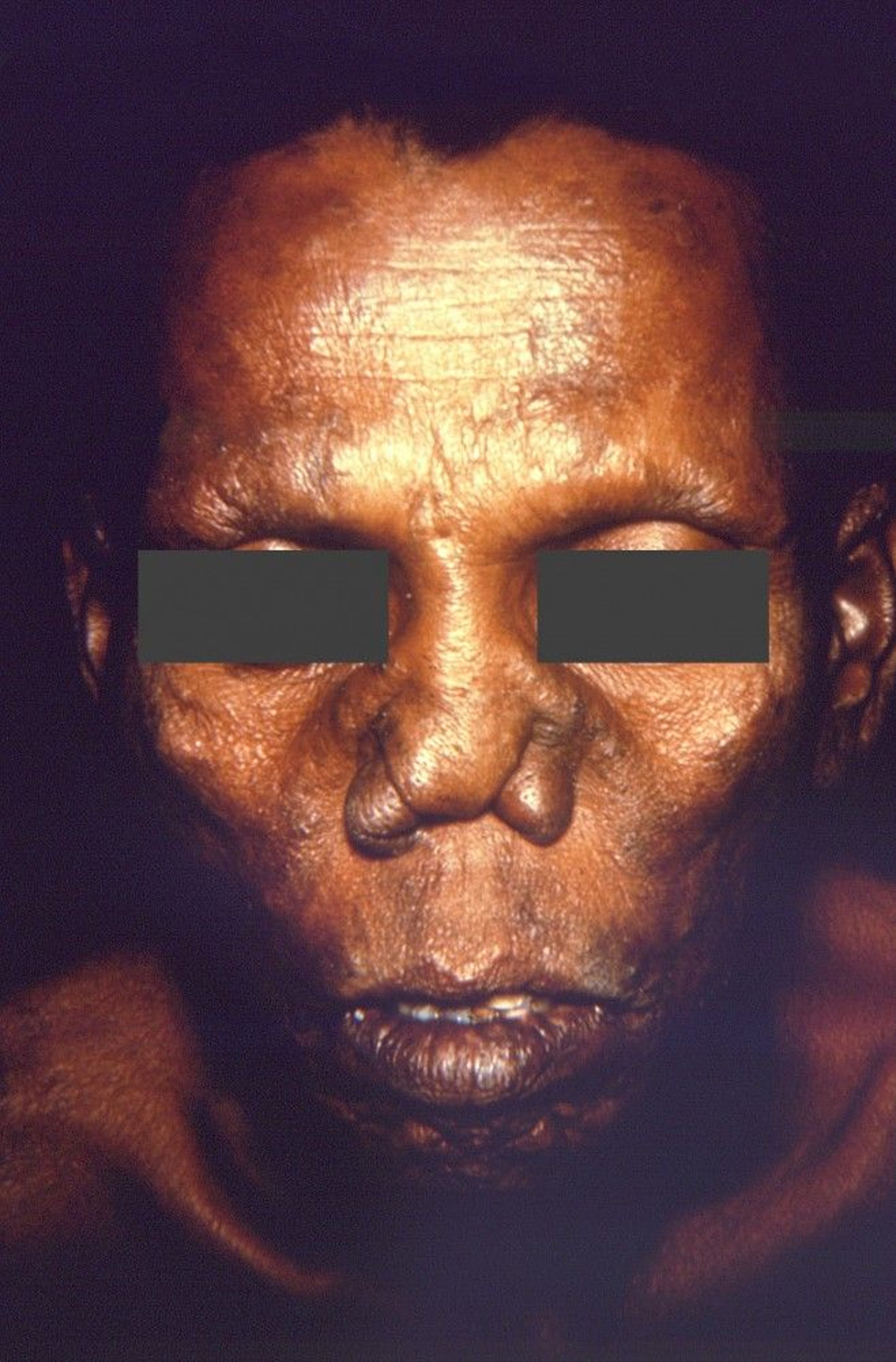
This photo shows a depressed nasal bridge due to disintegration of the nasal cartilage.
CDC
Other areas of the body may be affected:
Eyes: Iritis may lead to glaucoma, and corneal insensitivity may lead to scarring and blindness.
Nose: Damage to the nasal mucosa can result in chronic nasal congestion and nosebleeds and, if untreated, erosion and collapse of the nasal septum.
Feet: Plantar ulcers with secondary infection are a major cause of morbidity, making walking painful.
Kidneys:Amyloidosis and consequent kidney disease occasionally occur in lepromatous leprosy.
Sexual function: Men with lepromatous leprosy may have erectile dysfunction and infertility. The infection can reduce testosterone and sperm production by the testes.
Leprosy reactions
During the course of untreated or even treated leprosy, the immune system may produce inflammatory reactions. There are 2 types.
Type 1 leprosy reactions result from a spontaneous increase in cell-mediated immunity. These reactions can cause fever and inflammation of the preexisting skin and peripheral nerve lesions, resulting in skin edema, erythema, and tenderness and worsening nerve function. These reactions, particularly if not treated early, contribute significantly to nerve damage. Because the immune response is increased, these reactions are termed reversal reactions, despite the apparent clinical worsening.
Type 2 leprosy reactions (erythema nodosum leprosum, or ENL) are systemic inflammatory reactions that appear to be a vasculitis or panniculitis and probably involve circulating immune complex deposition or increased T-helper cell function. They have become less common since clofazimine was added to the drug regimen. Patients may develop erythematous and painful papules or nodules that may pustulate and ulcerate and cause fever, neuritis, lymphadenitis, orchitis, arthritis (particularly in large joints, usually knees), and glomerulonephritis. Hemolysis or bone marrow suppression may cause anemia, and hepatic inflammation may cause mild abnormalities in liver tests.(erythema nodosum leprosum, or ENL) are systemic inflammatory reactions that appear to be a vasculitis or panniculitis and probably involve circulating immune complex deposition or increased T-helper cell function. They have become less common since clofazimine was added to the drug regimen. Patients may develop erythematous and painful papules or nodules that may pustulate and ulcerate and cause fever, neuritis, lymphadenitis, orchitis, arthritis (particularly in large joints, usually knees), and glomerulonephritis. Hemolysis or bone marrow suppression may cause anemia, and hepatic inflammation may cause mild abnormalities in liver tests.
Diagnosis of Leprosy
Microscopic examination of skin biopsy specimen
Serology and molecular techniques
The diagnosis of leprosy is often delayed in the United States because clinicians are unfamiliar with the clinical manifestations.
Leprosy is suggested by the presence of skin lesions and peripheral neuropathy and confirmed by microscopic examination of biopsy specimens. M. leprae and M. lepromatosis do not grow on artificial culture media. Biopsy specimens should be taken from the advancing edge of tuberculoid lesions or, in lepromatous leprosy, from nodules or plaques.

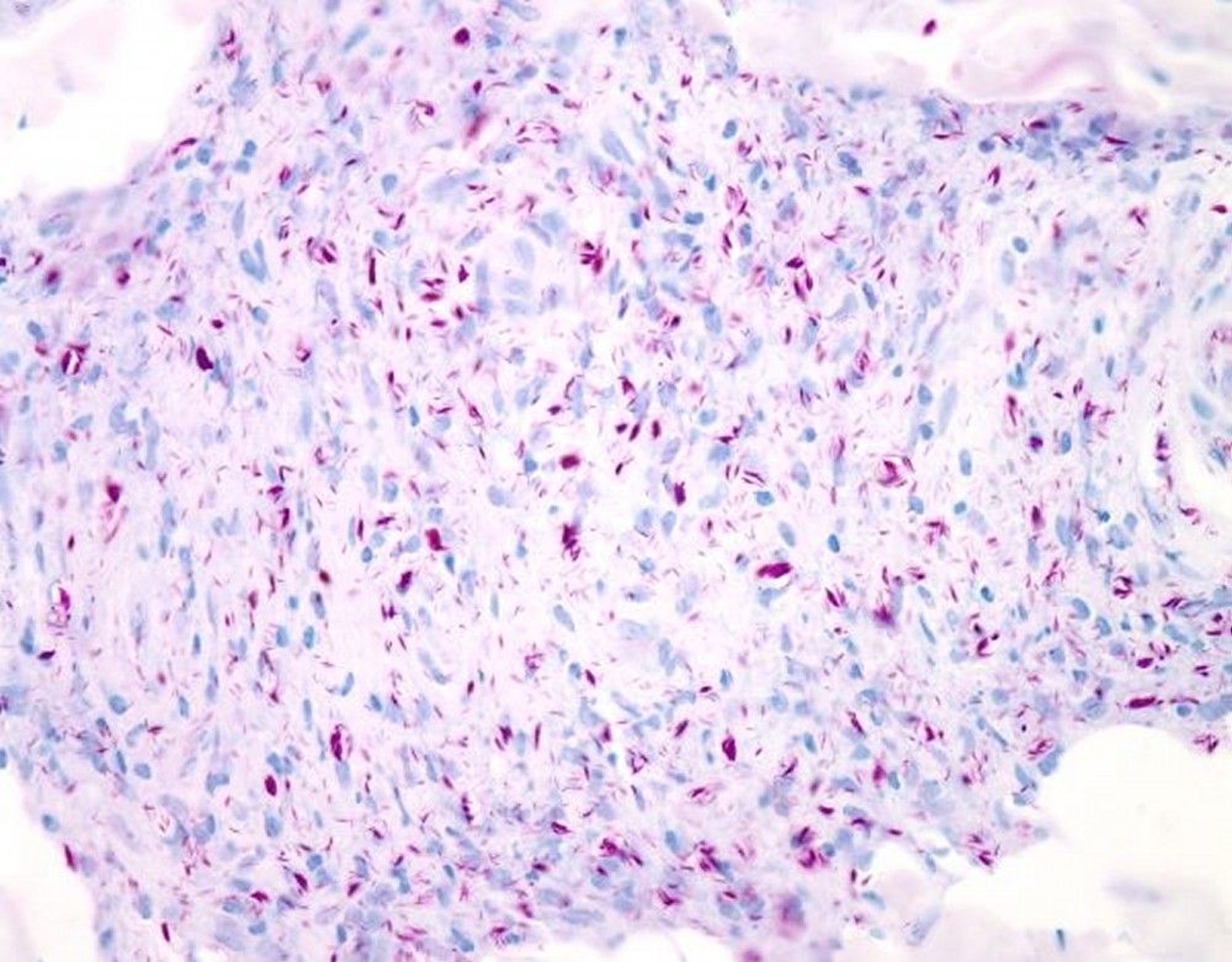
This image is a light micrograph of modified Ziehl-Neelsen–stained (Wade-Fite stain) M. leprae in a skin biopsy from a person with lepromatous leprosy. The mycobacteria appear red and are present in large numbers, singly as well as in clusters (globi). Magnification is 20X when printed at 10-cm wide.
WEBPATHOLOGY/SCIENCE PHOTO LIBRARY
Serologic tests, including serum IgM antibodies to M. leprae, are specific but insensitive (present in only two-thirds of patients with tuberculoid leprosy). Diagnostic usefulness is further limited in endemic areas because such antibodies may be present in asymptomatic infection.
Molecular techniques such as polymerase chain reaction (PCR) testing may aid diagnosis and are increasingly important, especially in patients with atypical or early disease (1).
Diagnosis reference
1. Mungroo MR, Khan NA, Siddiqui R. Mycobacterium leprae: Pathogenesis, diagnosis, and treatment options. Microb Pathog. 2020;149:104475. doi:10.1016/j.micpath.2020.104475
Treatment of Leprosy
Long-term, multidrug regimens with dapsone, rifampin (rifampicin), and clofazimineLong-term, multidrug regimens with dapsone, rifampin (rifampicin), and clofazimine
Antibiotics can stop the progression of leprosy but do not reverse any nerve damage or deformity. Thus, early detection and treatment are vitally important.
Because of antibiotic resistance, multidrug regimens are used. The medications chosen depend on the type of leprosy; multibacillary leprosy requires more intensive regimens and a longer treatment duration than paucibacillary leprosy does.
Advice about diagnosis and treatment is available from the Health Resources and Services Administration's National Hansen’s Disease (Leprosy) Program (or call 1-800-642-2477). Standard regimens recommended by the World Health Organization (WHO) differ somewhat from those used in the United States (1).
Medications for leprosy
DapsoneDapsone is relatively inexpensive and generally safe to use. Adverse effects include hemolysis and anemia (which are usually mild) and allergic dermatoses (which can be severe); rarely, dapsone syndrome (exfoliative dermatitis, high fever, mononucleosis-like white blood cell differential) occurs.
Rifampin (rifampicin),Rifampin (rifampicin), is primarily bactericidal for M. leprae and is even more effective than dapsone. It is available free from the WHO (2). Adverse effects include hepatotoxicity, flu-like syndromes, and, rarely, thrombocytopenia and renal failure.
ClofazimineClofazimine is extremely safe. The main adverse effect is reversible skin pigmentation, but discoloration may take months to resolve. Clofazimine can be obtained in the United States only from the Health Resources and Services Administration (3).
MinocyclineMinocycline is a tetracycline antibiotic with proven bactericidal activity against is a tetracycline antibiotic with proven bactericidal activity againstM. leprae.
MoxifloxacinMoxifloxacin is a fluoroquinolone with potent bactericidal activity against M. leprae; efficacy is comparable to rifampin.
Multibacillary leprosy
The standard WHO regimen includes dapsone, rifampin (rifampicin), and clofazimine. The WHO provides these medications free for all leprosy patients throughout the world (The standard WHO regimen includes dapsone, rifampin (rifampicin), and clofazimine. The WHO provides these medications free for all leprosy patients throughout the world (2). People with leprosy in the United States can also receive leprosy medications at no cost; however, some medications such as clofazimine require additional logistic considerations (on the part of the health care professional) before prescription (see also the National Hansen’s Disease (Leprosy) Program).
Per the WHO, adults with multibacillary disease should be administered the following regimen:
Rifampin (rifampicin) 600 mg orally once/month and clofazimine 300 mg orally once/month (with a health care professional’s supervision)Rifampin (rifampicin) 600 mg orally once/month and clofazimine 300 mg orally once/month (with a health care professional’s supervision)
Dapsone 100 mg orally once/day plus clofazimine 50 mg orally once/day (without supervision) (Dapsone 100 mg orally once/day plus clofazimine 50 mg orally once/day (without supervision) (1)
This regimen is continued for 12 months.
In the United States, the regimen is rifampin 600 mg orally once/month, moxifloxacin 400 mg orally once/month, and minocycline 100 mg orally once/month for 24 months. Clarithromycin 500 mg orally once/month or levofloxacin 500 mg orally once/month may be substituted for 1 of these medications (In the United States, the regimen is rifampin 600 mg orally once/month, moxifloxacin 400 mg orally once/month, and minocycline 100 mg orally once/month for 24 months. Clarithromycin 500 mg orally once/month or levofloxacin 500 mg orally once/month may be substituted for 1 of these medications (3).
Paucibacillary leprosy
Per the WHO, adults with paucibacillary disease should be administered the following regimen:
Rifampin (rifampicin) 600 mg orally once/month and clofazimine 300 mg orally once/month (with a health care professional’s supervision)Rifampin (rifampicin) 600 mg orally once/month and clofazimine 300 mg orally once/month (with a health care professional’s supervision)
Dapsone 100 mg orally once/day plus clofazimine 50 mg orally once/day (without supervision) (Dapsone 100 mg orally once/day plus clofazimine 50 mg orally once/day (without supervision) (1)
This regimen is continued for 6 months.
In the United States, the regimen is rifampin 600 mg orally once/month, moxifloxacin orally 400 mg once/month, and minocycline 100 mg orally once/month for 12 months. Clarithromycin 500 mg orally once/month or levofloxacin 500 mg orally once/month may be substituted for 1 of these medications (In the United States, the regimen is rifampin 600 mg orally once/month, moxifloxacin orally 400 mg once/month, and minocycline 100 mg orally once/month for 12 months. Clarithromycin 500 mg orally once/month or levofloxacin 500 mg orally once/month may be substituted for 1 of these medications (3).
Leprosy reactions
Patients with type 1 leprosy reactions (except minor skin inflammation) are given high-dose prednisone 40 to 60 mg orally once/day initially for 5 days, followed by low maintenance doses (often as low as 10 to 15 mg once/day) for 6 months (Patients with type 1 leprosy reactions (except minor skin inflammation) are given high-dose prednisone 40 to 60 mg orally once/day initially for 5 days, followed by low maintenance doses (often as low as 10 to 15 mg once/day) for 6 months (3). Minor skin inflammation does not need to be treated.
In the United States, low-dose methotrexate 7.5 mg to 10 mg orally once/week can be initiated for 6 months to minimize the dose of glucocorticoids (In the United States, low-dose methotrexate 7.5 mg to 10 mg orally once/week can be initiated for 6 months to minimize the dose of glucocorticoids (4). Low-dose prednisone 1 to 2.5 mg orally once/day may be added for 3 months before reassessment of the patient (). Low-dose prednisone 1 to 2.5 mg orally once/day may be added for 3 months before reassessment of the patient (5). Cyclosporine may be a useful second-line treatment for severe type 1 reactions in patients who do not respond to or who are unable to take glucocorticoids.). Cyclosporine may be a useful second-line treatment for severe type 1 reactions in patients who do not respond to or who are unable to take glucocorticoids.
Type 2 reactions are also called erythema nodosum leprosum reactions. First and second episodes of erythema nodosum leprosum may be treated, if mild, with aspirin or, if significant, with 1 week of prednisone 40 to 60 mg orally once/day plus antimicrobials. Type 2 reactions are also called erythema nodosum leprosum reactions. First and second episodes of erythema nodosum leprosum may be treated, if mild, with aspirin or, if significant, with 1 week of prednisone 40 to 60 mg orally once/day plus antimicrobials.
In the United States, treatment of type 2 reactions that are mild is similar to that of type 1 reactions (3). For recurrent or severe cases, thalidomide 100 to 300 mg orally once/day is the medication of choice. Adverse effects are mild constipation, mild leukopenia, and sedation. However, because of its teratogenicity, ). For recurrent or severe cases, thalidomide 100 to 300 mg orally once/day is the medication of choice. Adverse effects are mild constipation, mild leukopenia, and sedation. However, because of its teratogenicity,thalidomide should not be given to women who may become pregnant.
Treatment references
1. World Health Organization (WHO). Guidelines for the diagnosis, treatment and prevention of leprosy. 2018. Accessed August 26, 2025.
2. WHO. Leprosy (Hansen disease). Accessed October 3, 2025.
3. Health Resources and Services Administration. National Hansen's Disease (Leprosy) Program. August 2025. Accessed September 17, 2025.
4. Perez-Molina JA, Arce-Garcia O, Chamorro-Tojeiro S, et al. Use of methotrexate for leprosy reactions. Experience of a referral center and systematic review of the literature. Travel Med Infect Dis. 2020;37:101670. doi:10.1016/j.tmaid.2020.101670
5. Jesus JB, Sena CBC, Macchi BM, do Nascimento JLM. Cyclosporin A as an Alternative Neuroimmune Strategy to Control Neurites and Recover Neuronal Tissues in Leprosy. Neuroimmunomodulation. 2022;29(1):15-20. doi:10.1159/000517993
Prevention of Leprosy
Because leprosy is not very contagious, the risk of spread is low. Only the untreated lepromatous form is contagious, but, even then, the infection is not easily spread. However, household contacts (particularly children) of patients with leprosy should be monitored for development of symptoms and signs of leprosy. Once treatment has begun, leprosy cannot be spread.
The best prevention is:
Avoiding contact with bodily fluids (including respiratory droplets) from and the rash on infected people
The bacille Calmette-Guérin (BCG) vaccine, used to prevent tuberculosis (TB), provides some degree of protection against mycobacterial infections other than TB such as leprosy, but is not often primarily used for that purpose. The bacille Calmette-Guérin (BCG) vaccine, used to prevent tuberculosis (TB), provides some degree of protection against mycobacterial infections other than TB such as leprosy, but is not often primarily used for that purpose.
The WHO recommends a single dose of rifampin as preventive treatment for people ≥ 2 years of age who are contacts of leprosy patients (The WHO recommends a single dose of rifampin as preventive treatment for people ≥ 2 years of age who are contacts of leprosy patients (1). This treatment is given only after leprosy and TB disease have been ruled out and no other contraindications exist.
Prevention reference
1. World Health Organization (WHO). Guidelines for the diagnosis, treatment and prevention of leprosy. 2018. Accessed August 26, 2025.
Key Points
Leprosy is a chronic infection usually caused by the acid-fast bacilli Mycobacterium leprae.
Leprosy is not very contagious in untreated patients and not at all contagious once treatment starts.
Leprosy affects mainly the skin and peripheral nerves.
The most severe complications result from loss of the sense of touch, pain, and temperature; muscle weakness that can result in deformities; and disfiguring lesions of the skin and nasal mucosa.
Inflammatory reactions called leprosy reactions can occur and require treatment with glucocorticoids.
Diagnose based on molecular methods or biopsy; M. leprae and M. lepromatosis cannot be grown in culture.
Treatment involves multidrug regimens typically using dapsone, rifampin, and clofazimine.Treatment involves multidrug regimens typically using dapsone, rifampin, and clofazimine.
Drug Information for the Topic





