

Coarctation of the aorta is a localized narrowing of the aortic lumen that results in upper-extremity hypertension, left ventricular hypertrophy, and, if severe, malperfusion of the abdominal organs and lower extremities. Symptoms vary with the anomaly’s severity and range from headache, chest pain, cold extremities, fatigue, and leg claudication to fulminant heart failure and shock. A soft bruit may be heard over the coarctation site. Diagnosis is by echocardiography or by CT or MR angiography. Treatment is balloon angioplasty with stent placement, or surgical correction.
(See also Overview of Congenital Cardiovascular Anomalies.)
Coarctation of the aorta accounts for 6 to 8% of congenital heart anomalies. It occurs in 10 to 20% of patients with Turner syndrome. The male:female ratio is 2:1.
Pathophysiology of Coarctation of the Aorta
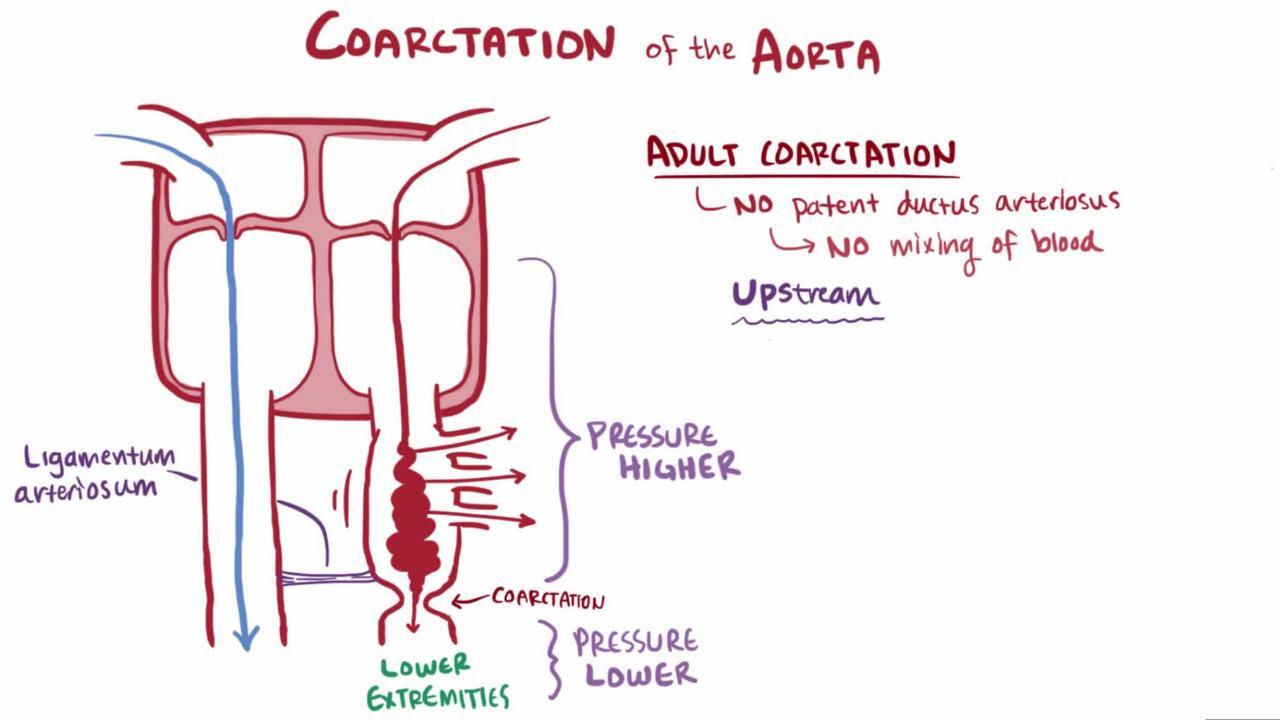
Coarctation of the aorta usually occurs at the proximal thoracic aorta just beyond the left subclavian artery and just across from the opening of the ductus arteriosus. Coarctation rarely involves the abdominal aorta. Thus, in utero and before the patent ductus arteriosus (PDA) closes, much of the cardiac output bypasses the coarctation via the PDA. Coarctation may occur alone or with various other congenital anomalies (eg, bicuspid aortic valve, ventricular septal defect, aortic stenosis, patent ductus arteriosus, mitral valve disorders, intracerebral aneurysms). The aneurysms associated with coarctation are usually saccular and are referred to as berry aneurysms.
Physiologic consequences involve 2 phenomena:
Pressure overload in the arterial circulation proximal to the coarctation
Hypoperfusion distal to the coarctation
Pressure overload causes left ventricular hypertrophy and hypertension in the upper part of the body, including the brain.
Hypoperfusion affects the abdominal organs and lower extremities. Malperfusion of the intestine increases the risk of sepsis due to enteric organisms.
Ultimately, the pressure gradient increases collateral circulation to the abdomen and lower extremities via intercostal, internal mammary, scapular, and other arteries.
Untreated coarctation may result in left ventricular hypertrophy, heart failure, collateral vessel formation, bacterial endocarditis, intracranial hemorrhage, hypertensive encephalopathy, and hypertensive cardiovascular disease during adulthood. Patients with untreated coarctation are at increased risk of aortic dissection or rupture later in life or in association with pregnancy. The ascending aorta is the area most frequently involved in dissection or rupture. Current data suggest that this risk is less likely a direct consequence of the coarctation and more likely related to a bicuspid aortic valve and associated aortopathy.
Symptoms and Signs of Coarctation of the Aorta
If coarctation is significant, circulatory shock with renal insufficiency (oliguria or anuria) and metabolic acidosis may develop within the first 7 to 10 days of life and may mimic findings of other systemic disorders such as sepsis. Infants with critical (severe) coarctation are likely to become acutely ill as soon as the ductus arteriosus constricts or closes.
Less severe coarctation may be asymptomatic during infancy. Subtle symptoms (eg, headache, chest pain, fatigue, and leg claudication during physical activities) may be present as children age. Upper-extremity hypertension is often present, but heart failure rarely develops after the neonatal period. Rarely, in adulthood, intracerebral aneurysms rupture, resulting in subarachnoid or intracerebral hemorrhage.
Typical physical examination findings include strong pulses and hypertension in the upper extremities, diminished or delayed femoral pulses, and a blood pressure (BP) gradient, with low or unobtainable arterial BP in the lower extremities. Since the coarctation site is usually adjacent to the ostium of the left subclavian artery, the left arm pulse and blood pressure may also be diminished, but they are still greater than leg pulses or blood pressures.
A grade 2 to 3/6 ejection systolic murmur is often present at the upper left sternal border, left axilla, and sometimes most prominently in the left interscapular area (see table ). An apical systolic ejection sound (click) may be present if a bicuspid aortic valve is also present. Dilated intercostal collateral arteries may cause a continuous murmur in the intercostal spaces.
Affected females may have Turner syndrome.

Diagnosis of Coarctation of the Aorta
Chest x-ray and ECG
Echocardiography or CT or MR angiography
Diagnosis is suggested by clinical examination (including BP measurement in all 4 extremities and comparing amplitude of brachial and femoral pulses), supported by chest x-ray and ECG, and established by 2-dimensional echocardiography with color flow and Doppler studies or, in older patients with a suboptimal echocardiographic window, with CT or MR angiography.
Chest x-ray shows coarctation as a “3” sign in the upper left mediastinal shadow. Heart size is normal unless heart failure supervenes. Dilated intercostal collateral arteries may erode the 3rd to 8th ribs, causing rib notching, but this radiographic sign is seldom seen before age 5 years.
ECG usually shows left ventricular hypertrophy but findings may be normal. Because the presence of a coarctation alters fetal circulation by shifting a higher proportion of flow through the right ventricle, neonates and infants with severe coarctation usually have right ventricular hypertrophy rather than left ventricular hypertrophy.
Treatment of Coarctation of the Aorta
For symptomatic neonates, prostaglandin E1 infusion
For hypertension, beta-blockers
Surgical correction for neonates, infants, and small children
Sometimes balloon angioplasty (sometimes with stent placement) in older children and adolescents
Symptomatic neonates are treated promptly. In infants with very mild coarctation and no signs of lower body hypoperfusion, the condition is monitored until definitive repair is done.
Medical management of coarctation
Symptomatic neonates require early surgical intervention, but preoperative cardiopulmonary stabilization is essential. Stabilization is done with infusion of prostaglandin E1 (beginning at 0.05 to 0.1 mcg/kg/minute) to reopen the constricted ductus arteriosus. Opening the ductus and its aortic ampulla provides some relief of the aortic obstruction. It also allows pulmonary artery blood to bypass the aortic obstruction via right-to-left shunting across the ductus and increases perfusion of the descending aorta, improving systemic perfusion and reversing metabolic acidosis.
Diuretics can help treat heart failure symptoms. IV milrinone can be useful in select circumstances (eg, infants with heart failure and significant left ventricle dysfunction).symptoms. IV milrinone can be useful in select circumstances (eg, infants with heart failure and significant left ventricle dysfunction).
In nonemergent situations, patients with hypertension may be treated with beta-blockers; angiotensin-converting enzyme (ACE) inhibitors may adversely affect renal function. After repair of the coarctation, hypertension may persist or develop years after repair and can be treated with beta-blockers, ACE inhibitors, angiotensin II receptor blockers, or calcium channel blockers.
Supplemental oxygen should be used with caution in neonates because the resulting decrease in pulmonary vascular resistance may increase pulmonary blood flow at the expense of systemic blood flow.
Coarctation repair
The preferred definitive treatment for coarctation remains somewhat controversial. Some centers prefer balloon angioplasty with or without stent placement; others prefer surgical correction and reserve the balloon procedure for recoarctation after surgical correction or for primary treatment of discrete coarctation in older children or adolescents. Both surgical and transcatheter techniques have generally good outcomes. Most studies have found a lower rate of periprocedural complications but a higher risk of restenosis and aneurysm after balloon angioplasty compared to surgical intervention.
Surgical options include
Resection and end-to-end anastomosis
Patch aortoplasty
Left subclavian flap aortoplasty
In severe coarctation manifesting early in life, the transverse aorta and isthmus are often hypoplastic, and this region of the aorta may need to be surgically enlarged.
Choice of surgical technique depends on anatomy and center preference. Surgical mortality rate is < 5% for symptomatic infants and < 1% for older children. Rarely, paraplegia results from ischemia to the spinal cord during cross-clamping of the aorta during surgery.
Balloon angioplasty is highly effective in treating recurrent coarctation after surgery.
Endocarditis prophylaxis is not needed preoperatively and is required only for the first 6 months after repair.
Key Points
Coarctation of the aorta is a localized narrowing of the lumen, typically in the proximal thoracic aorta just beyond the left subclavian artery and adjacent to the opening of the ductus arteriosus.
Manifestations depend on severity of coarctation but typically involve pressure overload proximal to the coarctation, leading to heart failure, and hypoperfusion distal to the coarctation.
Severe coarctation can manifest in the neonatal period with acidosis, renal insufficiency, and shock, but mild coarctation may not be apparent until an adolescent or adult is evaluated for hypertension or diminished femoral pulses.
There is typically a blood pressure gradient between upper and lower extremities, an easily distinguished amplitude differential between the upper and lower extremity pulses, and a grade 2 to 3/6 ejection systolic murmur, sometimes most prominent in the left interscapular area.
For symptomatic neonates, infuse prostaglandin E1 to reopen the constricted ductus arteriosus.
Correct coarctation surgically or using balloon angioplasty with or without stent placement.
More Information
The following English-language resources may be useful. Please note that THE MANUAL is not responsible for the content of these resources.
American Heart Association: Common Heart Defects: Provides overview of common congenital heart defects for parents and caregivers
American Heart Association: Infective Endocarditis: Provides an overview of infective endocarditis, including summarizing prophylactic antibiotic use, for patients and caregivers
Drug Information for the Topic





