

Osteogenesis imperfecta is a hereditary collagen disorder causing diffuse, abnormal fragility of bone and skeletal deformities. It is sometimes accompanied by sensorineural hearing loss, blue sclerae, dentinogenesis imperfecta, and joint hypermobility. Diagnosis is based on clinical manifestations and genetic testing. Treatment includes growth hormone for some types, bisphosphonates, and sometimes denosumab or teriparatide.
There are 5 clinical types of osteogenesis imperfecta (1–3):
I: Nondeforming with persistently blue sclerae
II: Perinatal lethal
III: Progressively deforming
IV: Moderate
V: With calcification of the interosseous membranes and/or hypertrophic callus
Their mode of inheritance is usually autosomal dominant, but types II, III, and IV have forms that are autosomal recessive. More than 85% of people who have one of these clinical types have mutations in the genes encoding the pro-alpha chains of type I procollagen (a structural component of bones, ligaments, and tendons): COL1A1 or COL1A2 (3).
There are a number of other, rarer types, which are caused by mutations in different genes.
General references
1. Marom R, Rabenhorst BM, Morello R. Osteogenesis imperfecta: an update on clinical features and therapies. Eur J Endocrinol. 2020;183(4):R95-R106. doi:10.1530/EJE-20-0299
2. Mortier GR, Cohn DH, Cormier-Daire V, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. 2019;179(12):2393-2419. doi:10.1002/ajmg.a.61366
3. Chaugule S, Constantinou CK, John AA, et al. Comprehensive Review of Osteogenesis Imperfecta: Current Treatments and Future Innovations. Hum Gene Ther. 2025;36(5-6):597-617. doi:10.1089/hum.2024.191
Symptoms and Signs of Osteogenesis Imperfecta
Hearing loss affects up to 70% of all patients with osteogenesis imperfecta and may occur in any of the 5 clinical types.
Type I (nondeforming with persistently blue sclera) is the mildest. Symptoms and signs in some patients are limited to blue sclerae (due to a deficiency in connective tissue allowing the underlying vessels to show through) and musculoskeletal pain due to joint hypermobility. Recurrent fractures in childhood are possible.
Type II (perinatal lethal or osteogenesis imperfecta congenita) is the most severe and is lethal. Multiple congenital fractures result in shortened extremities. Sclerae are blue. The skull is soft and, when palpated, feels like a bag of bones. Because the skull is soft, trauma during delivery may cause intracranial hemorrhage and stillbirth, or neonates may die suddenly during the first few days or weeks of life.

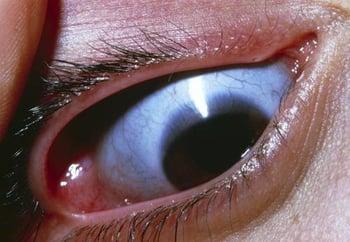
This photo shows a close-up of the eye showing a blue sclera (normally white).
Type III (progressively deforming) is the most severe nonlethal form of osteogenesis imperfecta. Patients with type III have short stature, spinal curvature, and multiple, recurrent fractures. Macrocephaly with triangular facies and pectal deformities are common. Scleral hue varies.
Cardiopulmonary abnormalities, including valvular disease, heart failure, and restrictive lung disease (both parenchymal and due to chest and spinal abnormalities), can be seen in types II and III.


This photo shows a patient with severe osteogenesis imperfecta who has a barrel chest, spinal curvature, severe bone deformities, loose joints, and poor muscle development.
Type IV (moderate) is intermediate/moderate in severity. Survival rate is high. This type is variable and deforming. Bones fracture easily in childhood before adolescence. Sclera are typically normal in color. Height is moderate-short stature. Accurate diagnosis is important because these patients may benefit from treatment.
Type V manifests with calcification of the interosseous membranes and/or hypertrophic callus and with radial head dislocation. Sclera are blue to bluish-gray. This type can be mild to moderate in severity and individuals with mild symptoms can lead relatively normal lives.
Diagnosis of Osteogenesis Imperfecta
Clinical evaluation
Analysis of type I procollagen and/or genetic testing
Sometimes prenatally with ultrasound, chorionic villus sampling, or amniocentesis
Diagnosis of osteogenesis imperfecta is usually clinical, but there are no standardized criteria.
Analysis of type I procollagen from cultured fibroblasts (from a skin biopsy) and/or sequencing of the COL1A1, COL1A2, and other causative genes is used to confirm the diagnosis.
Prenatally, osteogenesis imperfecta can be detected by detailed ultrasound; the optimal timing and likelihood of diagnosis depends on the type and severity (1). If a parental mutation is known, chorionic villus cells or amniocytes can be analyzed for that specific mutation. Chorionic villus cells (but not amniocytes) can also be cultured for analysis of the type I procollagen produced and for comparison with the procollagen test results of an affected family member.
Diagnosis reference
1. Byers PH, Krakow D, Nunes ME, Pepin M; American college of medical genetics. Genetic evaluation of suspected osteogenesis imperfecta (OI). Genet Med. 2006;8(6):383-388. doi:10.1097/01.gim.0000223557.54670.aa
Treatment of Osteogenesis Imperfecta
Bisphosphonates
Growth hormone
Sometimes denosumab or teriparatide
Sometimes vitamin D
Sometimes surgery
Treatment with bisphosphonates (eg, pamidronate 0.5 to 3 mg/kg IV once a day for 3 days, repeated as needed every 4 to 6 months; zoledronic acid 0.0125 to 0.025 mg/kg IV repeated every 3 to 6 months; alendronate 1 mg/kg [20 mg maximum] orally once a day) is aimed at increasing bone density and decreasing bone pain, and risk of fracture and scoliosis (Treatment with bisphosphonates (eg, pamidronate 0.5 to 3 mg/kg IV once a day for 3 days, repeated as needed every 4 to 6 months; zoledronic acid 0.0125 to 0.025 mg/kg IV repeated every 3 to 6 months; alendronate 1 mg/kg [20 mg maximum] orally once a day) is aimed at increasing bone density and decreasing bone pain, and risk of fracture and scoliosis (1, 2).
Growth hormone in conjunction with bisphosphonate therapy can improve growth and bone mineral density in some patients (2, 3).
Denosumab, a receptor activator of nuclear factor kappa-B ligand (RANKL), is a potent inhibitor of osteoclastic bone resorption and is typically given as an injection. Studies have shown that this medication is beneficial in some patients with osteogenesis imperfecta (Denosumab, a receptor activator of nuclear factor kappa-B ligand (RANKL), is a potent inhibitor of osteoclastic bone resorption and is typically given as an injection. Studies have shown that this medication is beneficial in some patients with osteogenesis imperfecta (2, 4).
Teriparatide, a recombinant parathyroid hormone, is used for 24 months and is given subcutaneously. It is not for use in children (Teriparatide, a recombinant parathyroid hormone, is used for 24 months and is given subcutaneously. It is not for use in children (5). Romosozumab, a sclerostin antibody, and fresolimumab, a transforming growth factor (TGF)-beta inhibitory antibody, are still in clinical trials (). Romosozumab, a sclerostin antibody, and fresolimumab, a transforming growth factor (TGF)-beta inhibitory antibody, are still in clinical trials (6).
Vitamin D supplementation should be provided to people who are deficient in this hormone.Vitamin D supplementation should be provided to people who are deficient in this hormone.
Orthopedic surgery, including implantation of rods into long bones, can stabilize growth and prevent fractures (2). Physical therapy and occupational therapy with a focus on sustainable muscle strength maintenance help improve function.
Cochlear implantation is indicated in selected cases of hearing loss.
Treatment references
1. Dwan K, Phillipi CA, Steiner RD, Basel D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev. 2016;10(10):CD005088, 2016. doi:10.1002/14651858.CD005088.pub4
2. Chaugule S, Constantinou CK, John AA, et al. Comprehensive Review of Osteogenesis Imperfecta: Current Treatments and Future Innovations. Hum Gene Ther. 2025;36(5-6):597-617. doi:10.1089/hum.2024.191
3. Antoniazzi F, Monti E, Venturi G, et al. GH in combination with bisphosphonate treatment in osteogenesis imperfecta. Eur J Endocrinol. 2010;163(3):479-487. doi:10.1530/EJE-10-0208
4. Li G, Jin Y, Levine MAH, et al. Systematic review of the effect of denosumab on children with osteogenesis imperfecta showed inconsistent findings. . Systematic review of the effect of denosumab on children with osteogenesis imperfecta showed inconsistent findings.Acta Paediatr. 2018;107(3):534–537. doi:10.1111/apa.14154
5. Liu W, Lee B, Nagamani SCS, et al. Approach to the Patient: Pharmacological Therapies for Fracture Risk Reduction in Adults With Osteogenesis Imperfecta. J Clin Endocrinol Metab. 2023;108(7):1787-1796. doi:10.1210/clinem/dgad035
6. Sun Y, Li L, Wang J, Liu H, Wang H. Emerging Landscape of Osteogenesis Imperfecta Pathogenesis and Therapeutic Approaches. ACS Pharmacol Transl Sci. 2024;7(1):72-96. Published 2024 Jan 2. doi:10.1021/acsptsci.3c00324
Drug Information for the Topic





