

Pulmonary Langerhans cell histiocytosis (PLCH) is proliferation of monoclonal Langerhans cells in lung interstitium and airspaces. Etiology is unknown, but cigarette smoking plays a primary role. Symptoms are dyspnea, cough, fatigue, and pleuritic chest pain. Diagnosis is based on history and imaging tests and sometimes on bronchoalveolar lavage and biopsy findings. Treatment is smoking cessation. Glucocorticoids are given in many cases, but efficacy is unknown. Lung transplantation is usually curative when combined with smoking cessation. Five-year survival is about 74%. Patients are at increased risk of cancer.
(See also Overview of Interstitial Lung Disease and Langerhans Cell Histiocytosis.)
Pulmonary Langerhans cell histiocytosis (PLCH) is a disease in which somatically mutated, monoclonal CD1a-positive Langerhans-like cells (a type of histiocyte or pulmonary antigen-presenting cell) infiltrate the bronchioles and alveolar interstitium, accompanied by lymphocytes, plasma cells, neutrophils, and eosinophils. It usually affects the middle and upper lobes of the lung. PLCH is one manifestation of Langerhans cell histiocytosis, which can affect many organs (most notably the lungs, skin, bones, pituitary, and lymph nodes) in isolation or simultaneously. PLCH occurs in isolation ≥ 85% of the time.
The etiology of PLCH is unknown, but the disease occurs almost exclusively in White people age 20 to 40 years who smoke (1). In other populations, PLCH is very rare, with one population-based study estimating its prevalence between 0.1 to 0.3/100,000 individuals per year (2). Men and women are affected equally. Women develop disease later, but differences in age at onset by sex may represent differences in smoking behavior. Pathophysiology may involve recruitment and proliferation of Langerhans cells in response to cytokines and growth factors secreted by alveolar macrophages in response to cigarette smoke.
General references
1. Mason RH, Foley NM, Branley HM, et al. Pulmonary Langerhans cell histiocytosis (PLCH): a new UK register. Thorax 2014;69(8):766-767. doi:10.1136/thoraxjnl-2013-204313
2. Watanabe R, Tatsumi K, Hashimoto S, Tamakoshi A, Kuriyama T; Respiratory Failure Research Group of Japan. Clinico-epidemiological features of pulmonary histiocytosis X. Intern Med 2001;40(10):998-1003. doi:10.2169/internalmedicine.40.998
Symptoms and Signs of PLCH
Typical symptoms and signs of pulmonary Langerhans cell histiocytosis are dyspnea, nonproductive cough, fatigue, fever, weight loss, and pleuritic chest pain. Sudden, spontaneous pneumothorax is common.
About 15% of patients have no symptoms, with disease noted incidentally on a chest radiograph taken for another reason.
Bone pain due to bone cysts (18%), rash (13%), and polyuria due to arginine vasopressin deficiency (5%) are the most common manifestations of extrapulmonary involvement and occur in up to 15% of patients, rarely being the presenting symptoms of PLCH. There are few signs of PLCH; the physical examination results are usually normal. In patients with progressive disease (worsening of diffusing capacity of the lung for carbon monoxide [DLCO] and evidence of worsening airway obstruction on pulmonary function testing), pulmonary hypertension may occur.
Diagnosis of PLCH
High-resolution CT (HRCT)
Pulmonary function tests
Sometimes bronchoscopy and biopsy
Pulmonary Langerhans cell histiocytosis is suspected based on history and chest radiograph findings and is confirmed by HRCT and bronchoscopy with biopsy and bronchoalveolar lavage.
Chest radiograph classically shows bilaterally symmetric nodular opacities in the middle and upper lung fields with cystic changes and normal or increased lung volumes. The lung bases are often spared. Appearance may mimic chronic obstructive pulmonary disease (COPD) or lymphangioleiomyomatosis.
Confirmation on HRCT of middle and upper lobe cysts (often with bizarre shapes) and/or nodules with interstitial thickening is considered diagnostic of PLCH.

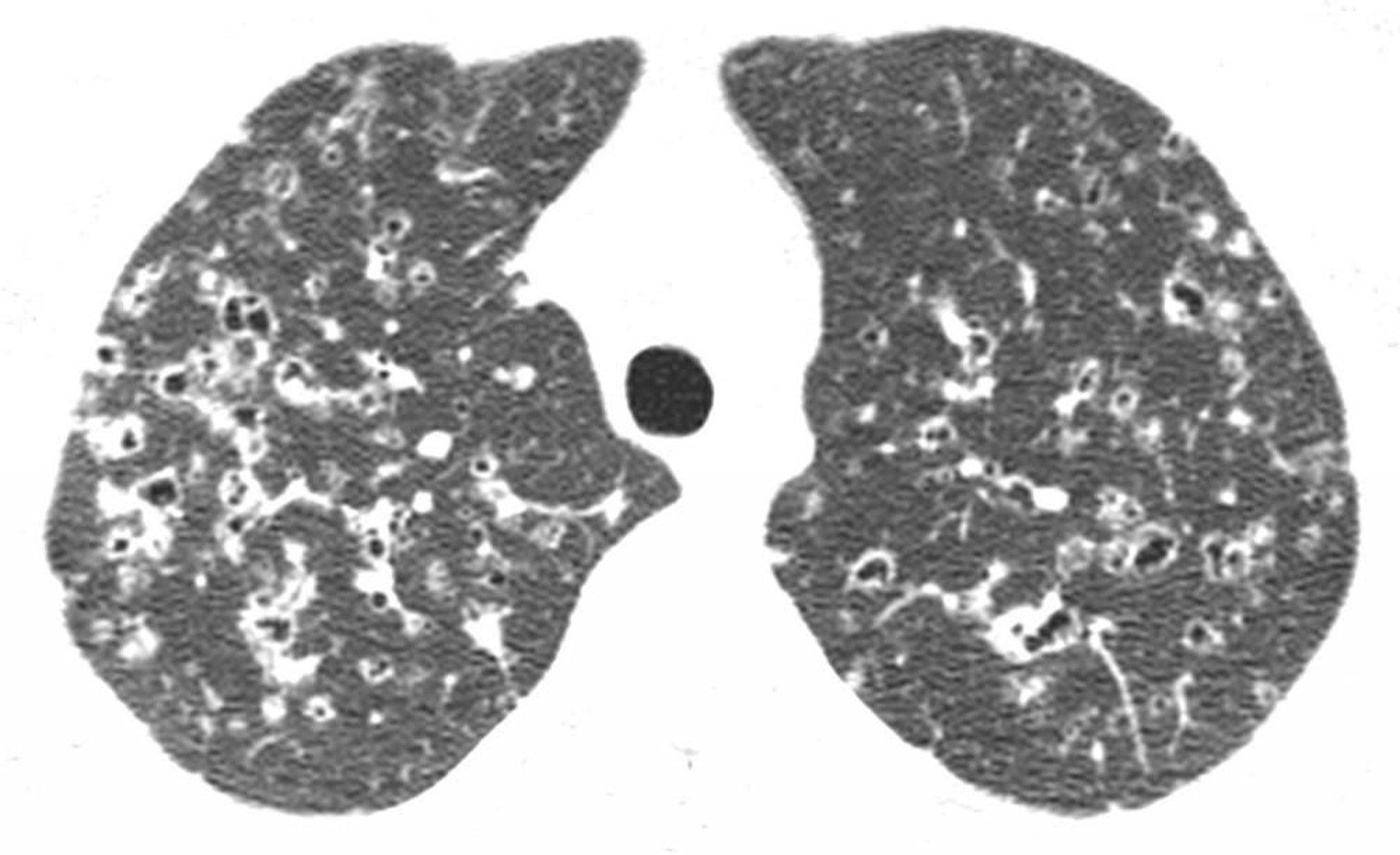
High-resolution CT through the upper lobes shows nodules and irregular air-density cysts, usually in the upper and mid-lung regions. These findings are characteristic of Langerhans cell histiocytosis in a patient with a history of smoking.
Image courtesy of Harold R. Collard, MD.
Pulmonary function test findings are normal, restrictive, obstructive, or mixed depending on when the test is done during the course of the disease. Most commonly, the DLCO is reduced and oxygenation during exercise is impaired.
Bronchoscopy and biopsy are indicated when history, imaging, and other test results are inconclusive. Finding > 5% of CD1a-positive cells in bronchoalveolar lavage fluid is highly suggestive of the disease when immunohistochemistry is performed; staining is usually positive for CD1a, S-100 protein, and CD207 (langerin). Biopsy shows proliferation of Langerhans cells with occasional clustering of eosinophils (the origin of the outdated term eosinophilic granuloma) in the midst of cellular and fibrotic nodules that may take on a stellate configuration.
Treatment of PLCH
Smoking cessation
Possibly glucocorticoids and cytotoxic drugs or lung transplantation
The main treatment of pulmonary Langerhans cell histiocytosis is smoking cessation, which leads to symptom resolution in up to one-third of patients.
Empiric use of glucocorticoids and cytotoxic drugs (eg, cladribine, cytarabine, vinblastine ) is common practice for patients with major pulmonary or extrapulmonary involvement (Empiric use of glucocorticoids and cytotoxic drugs (eg, cladribine, cytarabine, vinblastine ) is common practice for patients with major pulmonary or extrapulmonary involvement (1); however, evidence from large scale clinical trials is lacking.
Lung transplantation is an option for otherwise healthy patients with accelerating respiratory insufficiency, but the disorder may recur in the transplanted lung if the patient continues or resumes smoking.
Treatment reference
1. Elia D, Torre O, Cassandro R, Caminati A, Harari S. Pulmonary Langerhans cell histiocytosis: a comprehensive analysis of 40 patients and literature review. Eur J Intern Med 2015;26(5):351-356. doi:10.1016/j.ejim.2015.04.001
Prognosis for PLCH
Spontaneous resolution of symptoms occurs in some patients with minimally symptomatic pulmonary Langerhans cell histiocytosis. Overall, the 10-year survival is > 90% (1). The clinical markers for progressive disease include:
Continued smoking
Age extremes
Multiorgan involvement
Persistent constitutional symptoms
Numerous cysts on chest radiograph
Reduced DLCO
Low forced expiratory volume in 1 second (FEV1)/ forced vital capacity (FVC) ratio (< 66%)
High residual volume (RV)/total lung capacity (TLC) ratio (> 33%)
Need for prolonged corticosteroid use
Cause of death is respiratory insufficiency or cancer. Lung cancer risk is increased because of cigarette smoking.
Prognosis reference
1. Benattia A, Bugnet E, Walter-Petrich A, et al. Long-term outcomes of adult pulmonary Langerhans cell histiocytosis: a prospective cohort. Eur Respir J 2022;59(5):2101017. Published 2022 May 26. doi:10.1183/13993003.01017-2021
Key Points
In pulmonary Langerhans cell histiocytosis (PLCH), monoclonal Langerhans cells proliferate in the alveolar interstitium and bronchioles.
Consider PLCH in patients age 20 to 40 who smoke and in whom chest radiograph shows bilaterally symmetric nodular opacities in the middle and upper lung fields with cystic changes.
Confirm the diagnosis with high-resolution CT or, if results are inconclusive, lung biopsy.
Recommend smoking cessation.
Consider glucocorticoids and cytotoxic drugs and, if smoking has ceased and disease still persists, lung transplantation.
Drug Information for the Topic





