




El síndrome de Marfan se caracteriza por anomalías del tejido conectivo que provocan alteraciones oculares, esqueléticas y cardiovasculares (p. ej., dilatación de la aorta ascendente, que puede causar disección aórtica). El diagnóstico es clínico. El tratamiento puede incluir beta-bloqueantes profilácticos para retrasar la dilatación de la aorta descendente y cirugía aórtica profiláctica.
La herencia del síndrome de Marfan es autosómica dominante. El defecto molecular básico se debe a mutaciones del gen que codifica la glucoproteína fibrilina-1 (FBN1), que es el componente principal de las microfibrillas y ayuda a fijar las células a la matriz extracelular. El defecto estructural más importante afecta el aparato cardiovascular, el sistema musculoesquelético y los ojos. También hay compromiso pulmonar y del sistema nervioso central.
Se observan muchas manifestaciones diferentes de la mutación genética que causa síndrome de Marfan; sin embargo, éste suele reconocerse por la asociación de miembros largos, dilatación de la raíz aórtica y luxación del cristalino.
Signos y síntomas del síndrome de Marfan
Sistema cardiovascular
Los principales hallazgos son
Prolapso valvular
La mayoría de las complicaciones graves se deben a alteraciones patológicas de la raíz aórtica y la aorta ascendente. Se observa compromiso preferencial de la túnica media de la aorta en las zonas sometidas a máxima tensión hemodinámica. La aorta muestra dilatación progresiva o disección aguda, que comienza por los senos coronarios, a veces antes de los 10 años. La raíz aórtica se dilata en el 50% de los niños y en el 60-80% de los adultos, y puede provocar insuficiencia aórtica, en cuyo caso se puede auscular un soplo diastólico sobre la válvula aórtica.
Las cúspides y cuerdas tendinosas redundantes pueden conducir al prolapso o insuficiencia mitral; el prolapso mitral pueden producir un clic sistólico y un soplo telesistólico o, en casos graves, un soplo holosistólico.
Puede sobrevenir endocarditis bacteriana en las válvulas afectadas.
Sistema musculoesquelético
La gravedad es muy variable. Los pacientes son más altos que el promedio para la edad y la familia; el largo de ambos brazos supera la talla. La aracnodactilia (dedos delgados, desproporcionadamente largos) es notable, a menudo por el signo del pulgar (la falange distal del pulgar sobrepasa el borde del puño). La deformidad del esternón —pectus carinatum (desplazamiento hacia afuera) o tórax en embudo (desplazamiento hacia adentro)— es frecuente, así como la hiperextensibilidad articular (pero por lo general las pequeñas contracturas en flexión en los codos), el genu recurvatum (curvatura hacia atrás de las piernas en las rodillas), los pies planos, la cifoescoliosis y las hernias diafragmáticas e inguinales. Por lo general, el tejido subcutáneo es escaso. A menudo, hay paladar ojival.
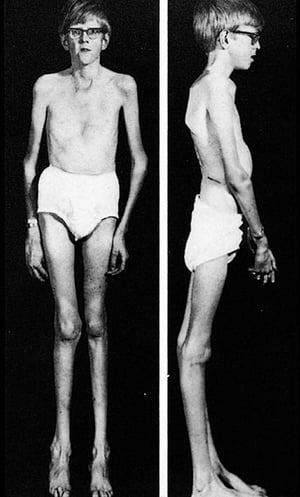
Con autorización del editor. De Macro R: Atlas of Heart Diseases: Congenital Heart Disease. Publicado por E Braunwald (editor) y RM Freedom. Philadelphia, Current Medicine, 1997.
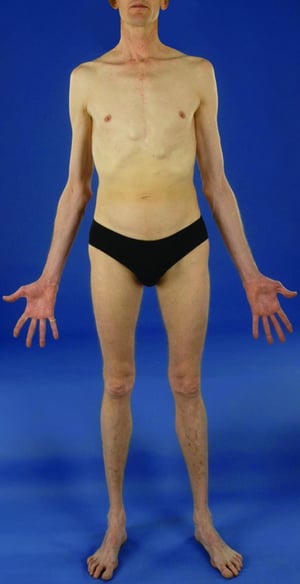
© Springer Science+Business Media

MEDICAL PHOTO NHS LOTHIAN/SCIENCE PHOTO LIBRARY

Foto cortesía de David D. Sherry, MD.

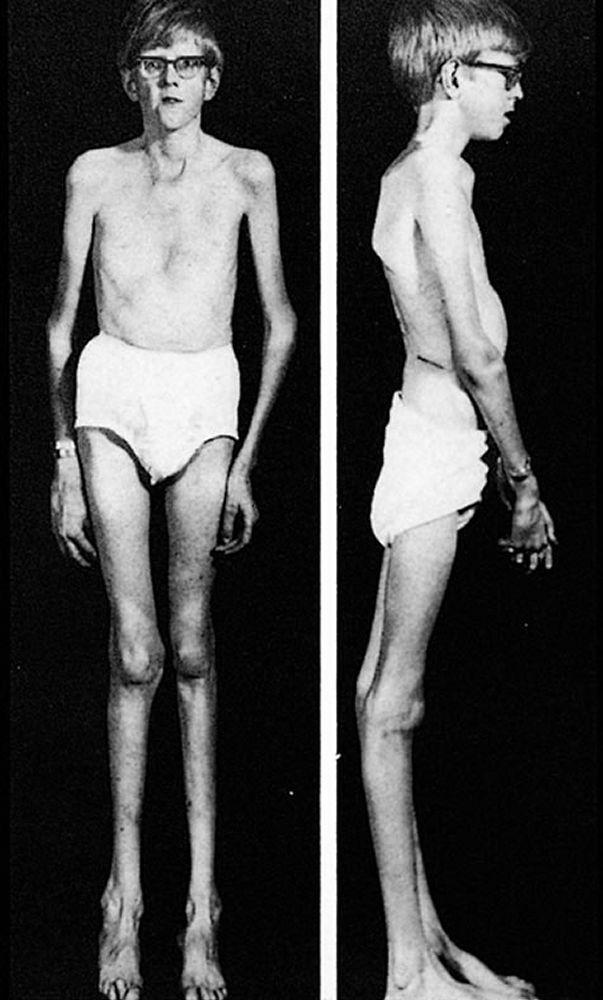
Con autorización del editor. De Macro R: Atlas of Heart Diseases: Congenital Heart Disease. Publicado por E Braunwald (editor) y RM Freedom. Philadelphia, Current Medicine, 1997.

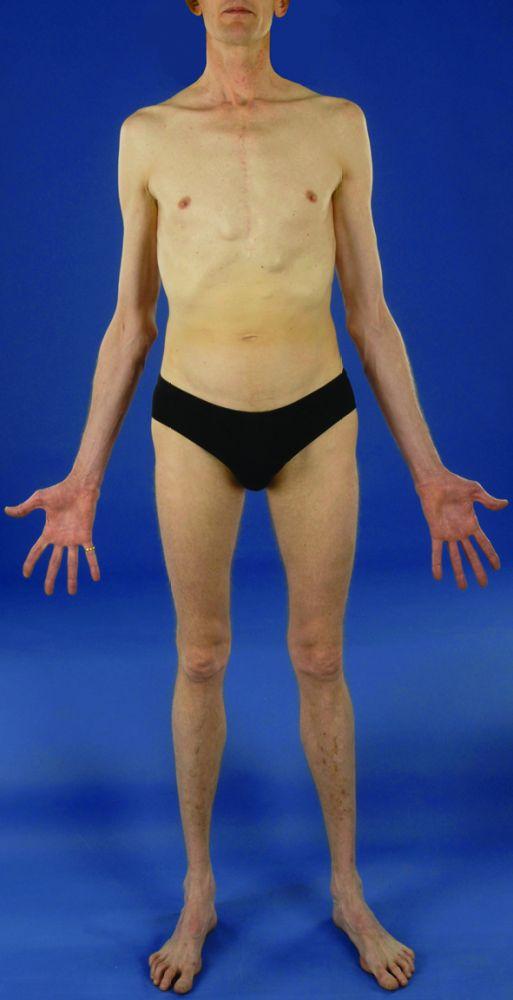
© Springer Science+Business Media

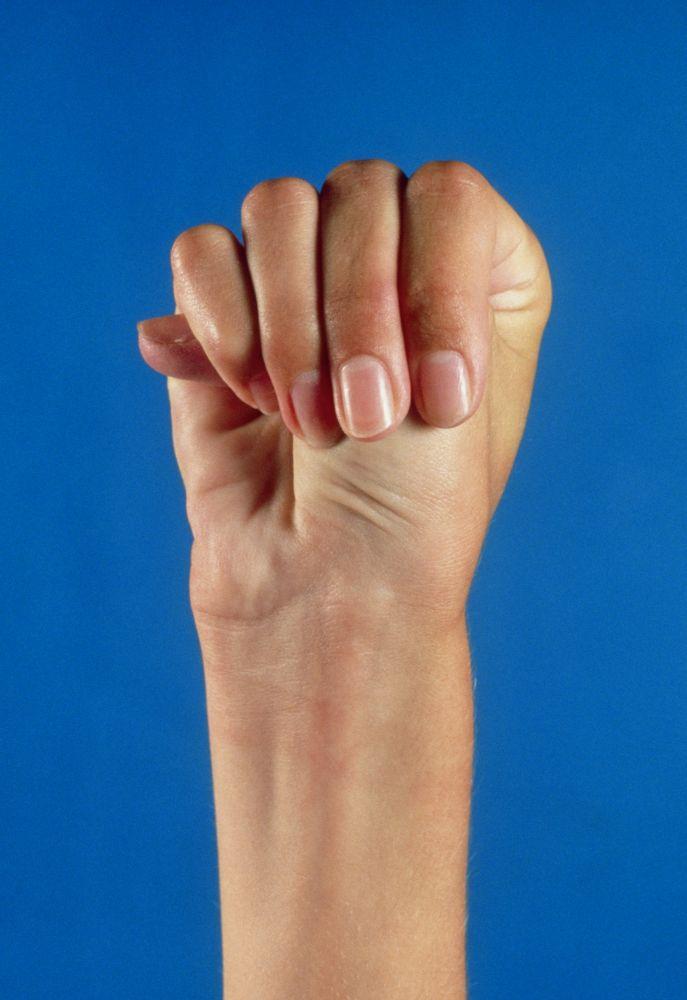
MEDICAL PHOTO NHS LOTHIAN/SCIENCE PHOTO LIBRARY

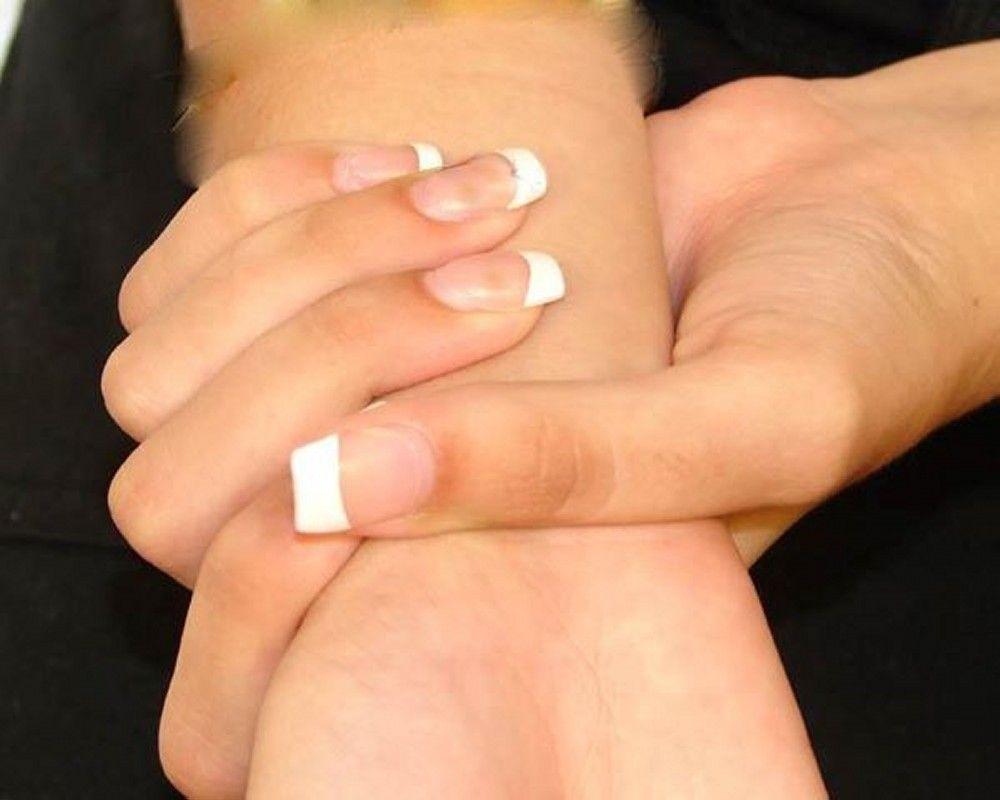
Foto cortesía de David D. Sherry, MD.
Ojos
Los hallazgos son ectopia del cristalino (subluxación o luxación hacia arriba del cristalino) e iridodonesis (temblor del iris). A menudo, puede observarse el borde del cristalino luxado a través de la pupila no dilatada. Puede haber miopía de alto grado, y puede ocurrir desprendimiento espontáneo de retina.
Aparato respiratorio
Es posible observar enfermedad pulmonar quística y neumotórax espontáneo recurrente. Estos trastornos pueden causar dolor y disnea.
Sistema nervioso central
La ectasia dural (ensanchamiento del saco dural que rodea a la médula espinal) es un hallazgo frecuente, y la mayoría de las veces se localiza en la columna lumbosacra. La ectasia dural puede causar cefalea, lumbalgia o déficits neurológicos manifestados por disfunción intestinal o vesical.
Diagnóstico del síndrome de Marfan
Criterios clínicos
Estudios genéticos
Ecocardiografía/RM (medición de la raíz aórtica, detección de prolapso valvular)
Examen con lámpara de hendidura (anomalías del cristalino)
Radiografías del sistema esquelético (mano, columna, tórax, pie y cráneo para investigar alteraciones características)
Resonancia magnética de la columna lumbosacra (ectasia dural)
El diagnóstico del síndrome de Marfan puede ser difícil, porque los pacientes tienen solo escasos signos y síntomas típicos y ninguna alteración histológica o bioquímica específica. Teniendo en cuenta esta variabilidad, los criterios diagnósticos se basan en constelaciones de hallazgos clínicos y antecedentes familiares y genéticos. (Para más información sobre el diagnóstico, véase la revised Ghent nosology [2010], que pondera en gran medida la presencia de dilatación o disección de la raíz aórtica y luxación del cristalino). No obstante, el diagnóstico es incierto en muchos casos parciales de síndrome de Marfan.
La homocistinuria puede remedar parcialmente este síndrome, pero es posible diferenciarla mediante la detección de homocistina en orina. Los estudios genéticos para las mutaciones FBN1 pueden ayudar a establecer el diagnóstico en personas que no cumplan con todos los criterios clínicos, pero existen casos con mutación del gen FBN1 negativa. La escasa correlación genotipo/fenotipo obstaculiza el diagnóstico prenatal por análisis de del gen FBN1 (se han descrito > 1.700 mutaciones diferentes).
Se realizan estudios por la imagen convencionales del sistema esquelético, el aparato cardiovascular y los ojos para detectar alguna anomalía estructural clínicamente relevante y aportar información que contribuya a los criterios diagnósticos (p. ej., ecocardiografía para identificar dilatación de la raíz aórtica).
Además de los criterios establecidos dentro de los aparatos y sistemas, se consideran criterios mayores los antecedentes familiares (familiar de primer grado con síndrome de Marfan) y genéticos (presencia de la mutación FBN1, causa conocida del síndrome de Marfan).
Pronóstico del síndrome de Marfan
Los avances terapéuticos y el control regular han mejorado la calidad de vida y reducido la mortalidad. La mediana de expectativa de vida aumentó de 48 años en 1972 a casi normal hoy en día en las personas que reciben atención médica apropiada. Sin embargo, la expectativa de vida todavía es menor en el paciente promedio, sobre todo debido a las complicaciones cardíacas y vasculares. Esta disminución puede imponer una carga emocional a un adolescente y su familia.
Tratamiento del síndrome de Marfan
Inducción de pubertad precoz en las niñas altas
Beta-bloqueantes
Reparación aórtica y valvular programada
Ortesis y cirugía en caso de escoliosis
El tratamiento del síndrome de Marfan se centra en la prevención y el manejo de las complicaciones.
En las niñas muy altas, la induccción de pubertad precoz a los 10 años de edad con estrógenos y progesterona puede reducir la talla adulta potencial.
Todos los pacientes deben recibir sistemáticamente beta-bloqueantes (p. ej., atenolol, propranolol) para ayudar a prevenir las complicaciones cardiovasculares. Estos fármacos reducen la contractilidad miocárdica y la presión diferencial y disminuyen la progresión de la dilatación aórtica y el riesgo de disección. También pueden administrarse bloqueantes del receptor de angiotensina II.
Se propone cirugía profiláctica si el diámetro aórtico es > 5 cm (menor en los niños). El riesgo de complicaciones aórticas es especialmente alto en las embarazadas; debe analizarse la reparación aórtica programada antes de la concepción. La insuficiencia valvular grave también se repara mediante cirugía. No está indicada la profilaxis de la endocarditis bacteriana antes de procedimientos invasivos, excepto en pacientes que tienen prótesis valvulares o con antecedentes de endocarditis infecciosa ( ver Procedimientos que requieren profilaxis antibiótica contra la endocarditis en pacientes con riesgo elevado en los Estados Unidos y ver Profilaxis recomendada contra la endocarditis durante procedimientos orodentales o en las vías respiratorias*).
La escoliosis se trata con ortesis durante el mayor tiempo posible, pero se recomienda intervención quirúrgica en pacientes con curvas de 40 a 50°.
Deben revaluarse anualmente los hallazgos cardiovasculares, esqueléticos y oculares (incluida la ecocardiografía).
Está indicado asesoramiento genético apropiado.
Conceptos clave
El síndrome de Marfan es el resultado de una mutación autosómica dominante del gen que codifica la glucoproteína fibrilina-1, que es el componente principal de las microfibrillas, lo que resulta en numerosas deformidades y defectos posibles.
Las manifestaciones varían ampliamente, pero los principales defectos estructurales afectan los sistemas cardiovascular, musculoesquelético y ocular, lo que produce una constelación típica de miembros largos, dilatación de la raíz aórtica, y cristalinos luxados.
La disección de la aorta es la complicación más peligrosa.
Diagnosticar mediante criterios clínicos; las pruebas genéticas se hacen a menudo.
Realizar pruebas de imágenes de los sistemas esquelético, cardiovascular y ocular para detectar anomalías estructurales.
Administrar a todos los pacientes un beta-bloqueante para ayudar a prevenir las complicaciones de la aorta; tratar otras complicaciones que puedan surgir.
Más información
El siguiente recurso en inglés puede ser útil. Tenga en cuenta que el MANUAL no es responsable por el contenido de este recurso.









