







L'amylose regroupe différentes pathologies qui ont pour point commun le dépôt extracellulaire de fibrilles insolubles composées de protéines mal agrégées. Le dépôt des protéines peut être soit localisé, responsable de peu de symptômes, soit diffus, affectant plusieurs organes et aboutissant à une défaillance multiviscérale. L'amylose peut être de novo ou secondaire à des maladies infectieuses, inflammatoires ou néoplasiques. Le diagnostic est établi par biopsie du tissu affecté; la protéine amyloïdogène est typée par diverses techniques immunohistologiques et biochimiques. Le traitement varie selon le type d'amylose.
Les fibrilles amyloïdes sont constituées de protéines normalement solubles mal repliées qui se regroupent en oligomères puis en fibrilles insolubles. Un certain nombre de protéines normales (type sauvage) et mutantes sont sensibles à un tel repliement et agrégation défectueux (protéines amyloïdogéniques), ce qui explique la grande variété des causes et des types d'amylose.
Les dépôts amyloïdes sont composés de petites fibrilles (environ 10 nm de diamètre), des fibrilles insolubles qui forment des feuillets congophiles bêta plissées qui peuvent être identifiés par diffraction des rayons X. Outre les protéines fibrillaires amyloïdes, les dépôts contiennent également le composant amyloïde P et des glycosaminoglycanes.
Les dépôts amyloïdes prennent une coloration rose à l'hématoxyline et à l'éosine, contiennent des constituants glucidiques qui sont colorés par le colorant acide périodique Schiff ou le bleu Alcian, mais de manière plus caractéristique ils ont une biréfringence vert pomme en microscopie en lumière polarisée après coloration au rouge Congo. À l'autopsie, les organes atteints apparaissent cireux.
Pour que l'amylose se développe en plus de la production de protéines amyloïdogéniques, il existe probablement aussi un dysfonctionnement des mécanismes normaux d'élimination de ces protéines mal repliées. Les dépôts amyloïdes sont métaboliquement inertes, mais perturbent de façon mécanique la structure et le fonctionnement de l'organe. Cependant, certains oligomères préfibrillaires de protéines amyloïdogéniques ont une toxicité cellulaire directe, une composante importante de la physiopathologie de la maladie.
Étiologie de l'amylose
Dans l'amyloïdose systémique, des protéines amyloïdogènes circulantes forment des dépôts dans divers organes. Les principaux types systémiques comprennent
AL (amylose primitive): causée par la surexpression acquise de chaînes légères d'immunoglobulines clonales
AF (amylose familiale): causée par la transmission d'un gène mutant codant pour une protéine sujette à un mauvais repliement, le plus souvent la transthyrétine (TTR)
L'ATTRwt (wild-type ATTR; précédemment appelée amyloïdose systémique sénile ou SSA [senile systemic amyloidosis]) causée par le mauvais repliement et l'agrégation de la TTR (transthyrétine) de type sauvage (wild type) (donc également appelée TTR)
AA (amylose secondaire): provoquée par l'agrégation de substances réactives de la phase aiguë, amyloïde sérique A
L'amylose provoquée par l'agrégation de bêta-2-microglobuline peut se produire chez les patients sous hémodialyse à long terme, mais l'incidence a diminué avec l'utilisation de membranes de dialyse modernes à haut débit. Il existe une forme héréditaire rare d'amylose à bêta-2-microglobuline due à une mutation du gène en cause.
Les formes localisées d'amylose semblent provoquées par la production locale et le dépôt d'une protéine amyloïdogène (le plus souvent les chaînes légères d'immunoglobulines) dans l'organe affecté, plutôt que par le dépôt de protéines en circulation. Les sites fréquemment impliqués comprennent le système nerveux central (p. ex., dans la maladie d'Alzheimer), la peau, les voies respiratoires supérieures ou inférieures, le parenchyme pulmonaire, la vessie, les yeux et les seins.
Amylose AL (amylose primitive)
L'AL est provoquée par la surproduction d'une chaîne légère d'immunoglobuline amyloïdogène chez des patients qui ont un trouble plasmocytaire monoclonal ou un autre trouble lymphoprolifératif des cellules B. Les chaînes légères peuvent également former des dépôts de tissus non fibrillaire (c'est-à-dire, maladie par dépôt de chaînes légères). Rarement, les chaînes lourdes d'immunoglobulines forment des fibrilles amyloïdes (appelée amyloïdose AH).
Les sites fréquents des dépôts amyloïdes sont la peau, les nerfs, le cœur, le tube digestif (dont la langue), les reins, le foie, la rate et les vaisseaux sanguins. Il existe habituellement une plasmocytose médullaire modérée similaire à celle du myélome multiple, bien que la plupart des patients n'aient pas de véritable myélome multiple (avec lésions osseuses lytiques, hypercalcémie, cylindres tubulaires rénaux et anémie). Cependant, environ 10 à 20% des patients présentant un myélome multiple développent également une amylose AL (light chain).
Amylose AF (amylose familiale)
L'amylose familiale est causée par l'héritage d'un gène codant pour une protéine sérique mutée à forte capacité d'agrégation, habituellement une protéine abondamment produite par le foie.
Les protéines sériques susceptibles de provoquer une amylose familiale comprennent la transthyrétine (TTR), l'apolipoprotéine A-I et A-II, le lysozyme, le fibrinogène, la gelsoline, et la cystatine C. Une forme supposée être familiale, est causée par le serum protein leukocyte chemotactic factor 2 (LECT2); cependant, la mutation spécifique du gène hérité de ce dernier type n'a pas été clairement démontrée.
L'amylose provoquée par la transthyrétine (ATTR) est le type le plus fréquent d'amylose familiale. Plus de 130 mutations du gène TTR ont été associées à l'amylose. La mutation la plus répandue, V30M, est fréquente au Portugal, en Suède, au Brésil et au Japon, et une mutation V122I est présente chez environ 4% des Noirs américains et des Caraïbes. La pénétrance de la maladie et l'âge d'apparition sont très variables, mais sont cohérentes au sein des familles et des groupes ethniques (1).
L'amylose ATTR (amylose provoquée par la transthyrétine) entraîne une neuropathie sensitivomotrice périphérique et végétative, une maladie rénale chronique et une cardiomyopathie. Un syndrome du canal carpien précède généralement d'autres manifestations de la maladie neurologique. Des dépôts vitrés peuvent se développer en raison de la production de TTR mutant par l'épithélium rétinien, ou bien des dépôts leptoméningés peuvent se développer à mesure que le plexus choroïde produit un TTR mutant. Lorsque la cardiomyopathie est la manifestation prédominante des dépôts de TTR dans le cœur, elle est appelée cardiomyopathie amyloïde par transthyrétine (ATTR-CM).
Amylose ATTRwt (amylose systémique sénile)
L'ATTRwt (wild-type ATTR; précédemment appelée amyloïdose systémique sénile ou SSA [senile systemic amyloidosis]) est provoquée par l'agrégation et le dépôt de TTR (transthyrétine) de type sauvage, principalement dans le cœur.
L'ATTRwt (wild-type ATTR; précédemment appelée amyloïdose systémique sénile ou SSA [senile systemic amyloidosis]) est de plus en plus reconnue comme une cause de la cardiomyopathie infiltrante chez l'homme âgé. Environ 16% des patients présentant une sténose aortique subissant un remplacement valvulaire aortique transcathéter (2) et 13% de ceux hospitalisés pour insuffisance cardiaque avec fraction d'éjection préservée ont également une cardiomyopathie amyloïde par transthyrétine, dans ce cas désignée comme wATTR-CM pour indiquer le dépôt de TTR de type sauvage dans le cœur (3). Les manifestations au niveau des tissus mous de l'amyloïde ATTRwt, dont le syndrome du canal carpien, la rupture du tendon bicipital, les déchirures de la coiffe des rotateurs et la sténose spinale, peuvent précéder de plusieurs années l'expression clinique de la cardiomyopathie infiltrante.
Les facteurs génétiques et épigénétiques menant à l'ATTRwt (wild-type ATTR; précédemment appelée amyloïdose systémique sénile ou SSA [senile systemic amyloidosis]) sont inconnus. Des patients souffrant d'ATTRwt (wild-type ATTR; précédemment appelée amyloïdose systémique sénile ou SSA [senile systemic amyloidosis]) et d'amylose AL (light chain), qui peuvent causer une cardiomyopathie et des gammapathies monoclonales amyloïdogènes, pouvant être présents chez les patients de ce groupe d'âge, il est essentiel de caractériser avec précision l'amyloïde afin que les patients qui ont une ATTRwt ne soient pas traités de manière inadéquate par la chimiothérapie (qui est utilisée dans l'amylose AL [light chain]).
Amylose AA (amylose secondaire)
Cette forme peut être secondaire à plusieurs pathologies infectieuses, inflammatoires ou malignes et elle est liée à une agrégation des isoformes des réactants de phase aiguë de l'amyloïde sérique.
Les infections responsables fréquentes sont les suivantes
Les pathologies inflammatoires prédisposantes comprennent les suivantes
Les syndromes de fièvre périodique héréditaires tels que la fièvre méditerranéenne familiale
Maladie de Castleman
Les cytokines inflammatoires (p. ex., l'interleukine (IL)-1, Tumor necrosis factor [TNF], IL-6) produites dans ces troubles ou de façon ectopique par les cellules tumorales augmentent la synthèse hépatique de l'amyloïde A sérique.
L'amylose AA (amyloïde A) touche préférentiellement les reins, la rate, le foie, les glandes surrénales et les ganglions lymphatiques. Les atteintes du cœur, ou nerfs périphériques et du système nerveux végétatif sont rares et surviennent tardivement dans l'évolution de la maladie.
Amylose localisée
L'amyloïdose localisée à l'extérieur du cerveau est le plus souvent causée par des dépôts de chaînes légères d'immunoglobuline clonales; dans le cerveau la protéine amyloïde bêta prédomine.
Les dépôts amyloïdes localisés touchent généralement les voies respiratoires et les tissus pulmonaires, la vessie et les uretères, la peau, les seins et les yeux. Plus rarement, d'autres protéines produites localement provoquent une amylose, telles que des isoformes kératiniques qui peuvent former des dépôts localement dans la peau. Les chaînes légères d'immunoglobuline clonales produites par le tissu lymphoïde associé à la muqueuse dans le tractus gastro-intestinal, les voies respiratoires et la vessie peuvent mener à une amylose AL (light chain) localisée dans ces organes.
Les dépôts de protéine amyloïde bêta dans le cerveau contribuent à la maladie d'Alzheimer ou à l'angiopathie amyloïde cérébrovasculaire. D'autres protéines produites dans le système nerveux central peuvent mal se replier, s'agréger, et léser les neurones, conduisant à des maladies neurodégénératives (p. ex., la maladie de Parkinson, la maladie de Huntington).
Références pour l'étiologie
1. Buxbaum JN, Ruberg FL: Transthyretin V122I (pV142I)* cardiac amyloidosis: an age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet Med 19(7):733-742, 2017. doi:10.1038/gim.2016.200
2. Fabbri G, Serenelli M, Cantone A, et al: Transthyretin amyloidosis in aortic stenosis: clinical and therapeutic implications. Eur Heart J Suppl 23(Suppl E):E128-E132, 2021. doi:10.1093/eurheartj/suab107
3. Magdi M, Mostafa MR, Abusnina W, et al: A systematic review and meta-analysis of the prevalence of transthyretin amyloidosis in heart failure with preserved ejection fraction. Am J Cardiovasc Dis 12(3):102-111, 2022. PMID: 35873185
Symptomatologie de l'amylose
La symptomatologie de l'amylose systémique n'est pas spécifique, entraînant souvent des retards diagnostiques. La suspicion d'amylose doit être augmentée chez les patients qui ont une maladie multisystémique évolutive.
Des dépôts amyloïdes rénaux se produisent généralement dans la membrane glomérulaire conduisant à une protéinurie, mais dans environ 15% des cas, les tubules sont touchés, provoquant une azotémie avec protéinurie minime. Ces processus peuvent évoluer vers le syndrome néphrotique avec hypoalbuminémie marquée, œdème et anasarque ou vers l'insuffisance rénale terminale.
L'atteinte hépatique se traduit par une hépatomégalie indolore, pouvant être majeure. Le bilan hépatique est généralement en faveur d'une cholestase intrahépatique avec élévation des phosphatases alcalines et plus tard de la bilirubine, bien que l'ictère soit rare. Parfois, une hypertension portale apparaît avec des varices œsophagiennes et une ascite.
L'atteinte des voies respiratoires et laryngées induit une dyspnée, une raucité de la voix, un wheezing, une hémoptysie, ou une obstruction des voies respiratoires.
L'infiltration du myocarde provoque une cardiomyopathie restrictive, pour aboutir finalement à une dysfonction diastolique et à une insuffisance cardiaque; un bloc cardiaque ou une arythmie peuvent se produire. L'hypotension est fréquente.
Une neuropathie périphérique, avec des paresthésies des doigts et des orteils, est un mode de présentation fréquent des amyloses AL et ATTR (amylose provoquée par la transthyrétine). La neuropathie végétative peut entraîner une hypotension orthostatique, des troubles de l'érection, des anomalies de la sudation, rétention urinaire et des troubles de la motilité du tube digestif.
L'angiopathie amyloïde cérébrovasculaire provoque le plus souvent une hémorragie cérébrale spontanée mais certains patients présentent des symptômes neurologiques brefs et transitoires.
L'atteinte amyloïde du tube digestif entraîne des anomalies de la motilité de l'œsophage ainsi que de l'intestin grêle et du colon. Une atonie gastrique, une malabsorption, une hémorragie ou une pseudo-occlusion peuvent également survenir. La macroglossie est une des manifestations de l'amylose AL (light chain).
L'amyloïdose des tissus mous précède typiquement l'expression clinique de la cardiomyopathie amyloïde ATTRwt. Les manifestations de la maladie amyloïde des tissus mous comprennent le syndrome du canal carpien, le doigt à ressort, la rupture du tendon bicipital et la sténose spinale.

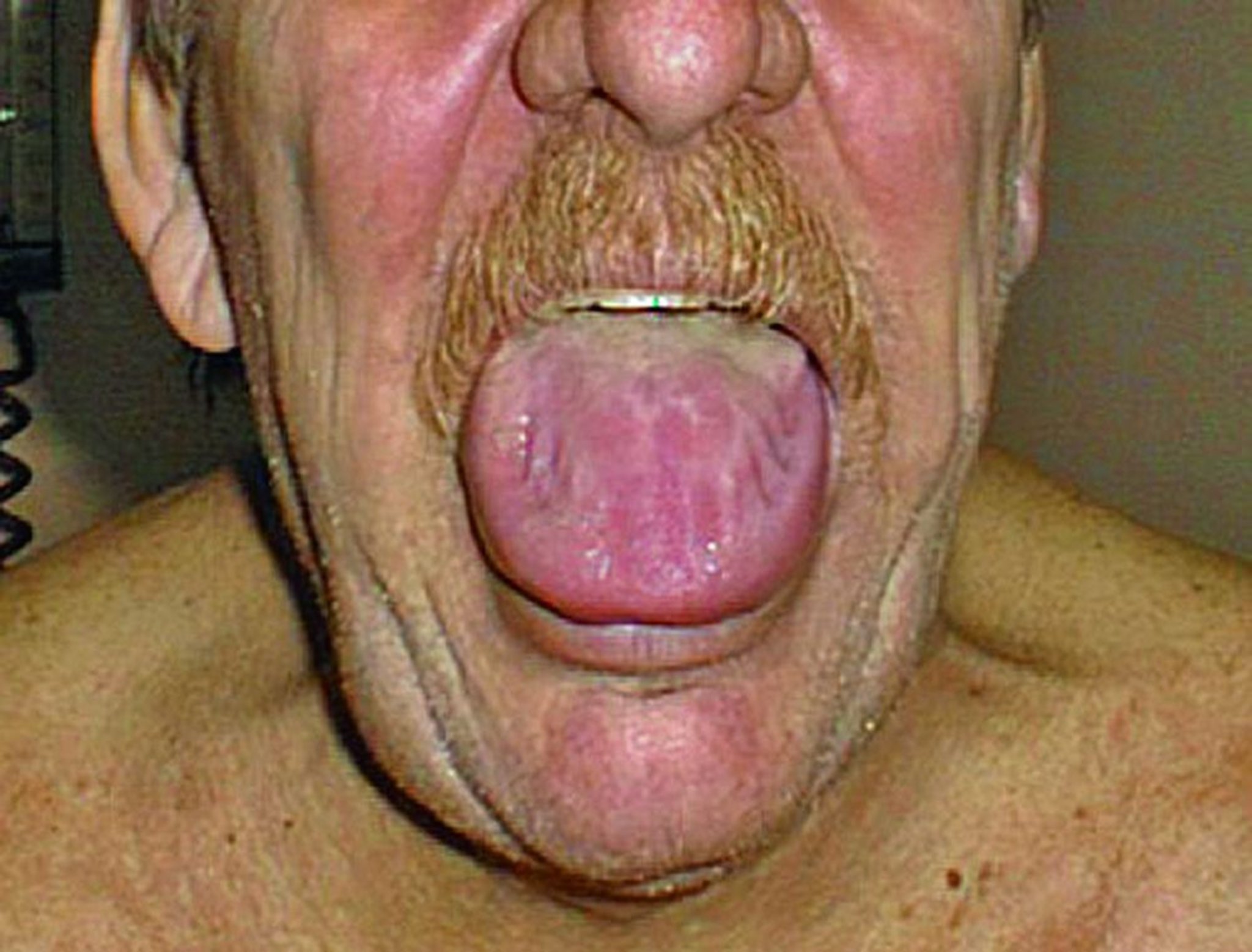
© Springer Science+Business Media
L'amyloïdose de la glande thyroïde peut provoquer la formation d'un goitre ferme, symétrique, non douloureux ressemblant à celui de la thyroïdite d'Hashimoto. D'autres endocrinopathies peuvent également se produire.
L'atteinte pulmonaire (principalement dans l'amylose AL) peut être caractérisée par des nodules pulmonaires et des kystes focaux, des lésions trachéobronchiques, des épanchements pleuraux ou des dépôts alvéolaires-septaux (interstitiels) diffus.
Des opacités vitréennes amyloïdes et des marges pupillaires festonnées bilatérales se développent dans plusieurs amyloses héréditaires.
D'autres manifestations comprennent des ecchymoses, souvent autour des yeux (yeux de raton laveur), causées par des dépôts amyloïdes dans les vaisseaux sanguins. Les dépôts amyloïdes provoquent l'affaiblissement des vaisseaux sanguins, qui peuvent se rompre à l'occasion d'un traumatisme mineur tel que des éternuements ou de la toux.
Diagnostic de l'amylose
Biopsie
Détermination du type d'amyloïde
Examen pour l'atteinte des organes
Biopsie
Le diagnostic d'amylose est fait par la démonstration de dépôts fibrillaires dans un organe impliqué. L'aspiration de la graisse abdominale sous-cutanée détecte les dépôts amyloïdes chez environ 80% des patients qui ont une amylose AL, mais dans moins de 25% des patients qui ont une ATTRwt (wild-type ATTR) (1). Si le résultat de la biopsie du tissu adipeux est négatif, un organe cliniquement atteint doit être biopsié. La sensibilité diagnostique des biopsies rénales et cardiaques est presque de 100% lorsque ces organes sont cliniquement atteints. Les coupes de tissus sont colorées au rouge Congo et examinées en lumière polarisée afin de mettre en évidence la biréfringence caractéristique. Des fibrilles de 10 nm non divergentes peuvent également être reconnues en microscopie électronique sur les biopsies cardiaques ou rénales.
La scintigraphie utilisant des traceurs qui se fixent sur l'os permet de diagnostiquer la cardiomyopathie amyloïde ATTR sans biopsie cardiaque, à condition que l'amylose AL ait été exclue.
Détermination du type d'amyloïde
Après que l'amylose aura été confirmée par biopsie, le type est déterminé par une variété de techniques. Dans certains types d'amylose, l'immunohistochimie ou l'immunofluorescence peuvent être diagnostiques, mais des résultats faux positifs sont possibles. D'autres techniques utiles comprennent le séquençage des gènes dans le cas de l'amylose familiale et l'identification biochimique précise par spectrométrie de masse des variants protéiques des dépôts amyloïdes (la méthode la plus sensible et la plus spécifique).
Si une amylose AL (light chain) est suspectée, les patients doivent être évalués à la recherche d'un trouble plasmocytaire sous-jacent en effectuant une mesure quantitative des chaînes légères d'immunoglobulines sériques libres, une détection qualitative sérique ou urinaire des chaînes légères monoclonales par électrophorèse avec immunofixation (l'électrophorèse des protéines sériques et l'électrophorèse des protéines urinaires sont peu sensibles dans le cas des patients atteints d'amylose AL [light chain]), et une biopsie de la moelle osseuse avec cytométrie en flux ou une immunohistochimie pour établir la clonalité des plasmocytes.
Les patients qui ont des plasmocytes clonaux de > 10% doivent être testés pour vérifier s'ils répondent aux critères du myélome multiple, avec dépistage de lésions osseuses lytiques, de l'anémie, de l'insuffisance rénale, et de l'hypercalcémie.
Atteinte d'organes
Les patients sont examinés à la recherche d'une atteinte d'organes en commençant par les tests non invasifs:
Reins: analyse d'urine; mesure de l'urée sérique, de la créatinine et de l'albumine sériques; taux de filtration glomérulaire estimé (eGFR); et collecte d'urine de 24 heures pour l'électrophorèse des protéines (UPEP)
Foie: bilan hépatique
Poumons: rx thorax, TDM du thorax et épreuves fonctionnelles respiratoires
Cœur: ECG et mesure de biomarqueurs tels que le brain (B-type) natriuretic peptide (BNP) sérique ou le N-terminal pro-BNP (NT-pro-BNP) et de la troponine
L'atteinte cardiaque peut être suggérée par un hypovoltage sur l'ECG (provoqué par un épaississement du ventricule), et/ou des troubles du rythme. Si une atteinte cardiaque est suspectée en raison de symptômes, en plus des signes ECG et des biomarqueurs cardiaques, une échocardiographie est effectuée pour mesurer la relaxation diastolique et la déformation longitudinale globale (une mesure de la fonction systolique ventriculaire gauche) et pour dépister une hypertrophie biventriculaire. Dans les cas ambigus, une IRM cardiaque peut être effectuée pour détecter un rehaussement au gadolinium sous-endocardique persistant, un signe caractéristique. Les scintigraphies cardiaques au pyrophosphate de technétium améliorent la détection de la cardiopathie amyloïde ATTR et peuvent éviter de recourir à des biopsies cardiaques à condition que les examens sanguins excluent l'amylose AL (2, 3).
Références pour le diagnostic
1. Aimo A, Emdin M, Musetti V, et al: Abdominal Fat Biopsy for the Diagnosis of Cardiac Amyloidosis. JACC Case Rep 2(8):1182-1185, 2020. doi:10.1016/j.jaccas.2020.05.062
2. Gillmore JD, Maurer MS, Falk RH, et al: Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 133(24):2404–2412, 2016.
3. Maurer MS, Bokhari S, Damy T, et al: Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail 12(9):e006075, 2019.
Traitement de l'amylose
Soins de support
Traitement spécifique de type
Il existe des traitements spécifiques pour la plupart des formes d'amylose, bien que certains traitements soient expérimentaux. Pour toutes les formes d'amylose systémique, les soins de soutien peuvent soulager les symptômes et améliorer la qualité de vie.
Soins de support
Les soins de support sont dirigés vers le système d'organe atteint:
Rein: les patients qui ont un syndrome néphrotique et un œdème doivent être traités par restriction sodée et hydrique, et des diurétiques de l'anse; en raison d'une perte de protéines, l'apport en protéines ne doit pas être limité. La transplantation rénale est une option lorsque le processus pathologique sous-jacent est contrôlé et peut permettre une survie à long terme comparable à celle d'autres maladies rénales.
Cœur: les patients qui ont une cardiomyopathie doivent être traités par restriction sodée et hydrique et des diurétiques de l'anse. D'autres médicaments de l'insuffisance cardiaque, dont la digoxine, les inhibiteurs de l'ECA, les inhibiteurs calciques, et les bêta-bloqueurs sont mal tolérés et contre-indiqués. La transplantation cardiaque a été pratiquée avec succès chez des patients soigneusement sélectionnés, atteints d'amylose AL (light chain) ou ATTR avec atteinte cardiaque grave. Pour prévenir les récidives sur le cœur greffé, les patients qui ont une amylose AL (light chain) doivent recevoir une chimiothérapie agressive dirigée contre le trouble plasmocytaire clonal et chez les patients présentant une polyneuropathie amyloïde ou une cardiomyopathie symptomatique on doit envisager d'utiliser les thérapies anti-ATTR.
Digestif: les patients atteints de diarrhée peuvent tirer profit du lopéramide. En cas de satiété précoce et de rétention gastrique le métoclopramide peut être utile.
Système nerveux: les douleurs des patients qui ont une neuropathie périphérique, peuvent être réduites par la gabapentine, la prégabaline ou la duloxétine.
L'hypotension orthostatique s'améliore souvent avec des doses élevées de la midodrine; ce médicament peut causer une rétention urinaire chez les hommes âgés, mais la complication médicamenteuse de l'hypertension de décubitus est rarement un problème dans cette population. Les bas de contention peuvent également être utiles, et la fludrocortisone peut être utilisée chez le patient qui n'a pas d'œdème périphérique, d'anasarque ou d'insuffisance cardiaque. En cas d'hypotension orthostatique réfractaire, de la midodrine, de la fludrocortisone ou de la droxidopa peuvent être ajoutées.
Amylose AL
Pour l'amylose AL:
Il est essentiel de débuter rapidement la thérapie anti-plasmocytaire pour préserver la fonction des organes et prolonger la vie.
La plupart des médicaments utilisés pour le myélome multiple ont été utilisés dans l'amylose AL; le choix du médicament, la dose et le calendrier doivent souvent être modifiés lorsque la fonction des organes est altérée.
La chimiothérapie par un agent alkylant (p. ex., le melphalan, le cyclophosphamide) associée à des corticostéroïdes a été le premier traitement faire apparaître un bénéfice. Le melphalan IV à haute dose associé à une autogreffe de cellules souches peut être très efficace chez des patients sélectionnés (1).
Les inhibiteurs du protéasome (p. ex., le bortézomib) et les immunomodulateurs (p. ex., le lénalidomide) peuvent également être efficaces. Un essai de l'anticorps monoclonal daratumumab plus cyclophosphamide, du bortézomib et de la dexaméthasone chez les patients qui ont une amylose AL récemment diagnostiquée (à l'exception des sujets qui ont une insuffisance cardiaque de classe NYHA III et IV, une protéine natriurétique N-terminale pro-B [NTproBNP] > 8500 pg/mL [> 1005 pmol/L] et un taux de filtration glomérulaire estimé à < 20 mL/minute/m2) ont montré un taux élevé de réponse hématologique sans précédent (2). La réponse hématologique est basée sur les taux sériques et urinaires de la protéine monoclonale déterminés par électrophorèse avec immunofixation et sur des taux de chaînes légères sériques avec des rapports kappa/lambda. Cependant, les données de survie à long terme font défaut.
Tous les traitements disponibles ciblent les lymphocytes B ou les plasmocytes dans l'amylose AL. Des études sur les anticorps anti-fibrilles, tels que le birtamimab et CAEL-101, sont en cours (3).
L'amylose AL localisée peut être traitée par radiothérapie externe à faible dose car les plasmocytes sont très radiosensibles.
Amylose ATTR
Pour l'amylose ATTR:
Transplantation hépatique
Médicaments stabilisants du tétramer
Médicament de blocage des gènes
La transplantation hépatique qui remplace le site primitif de synthèse de la protéine mutante par un nouvel organe qui produit de la TTR normale, peut être efficace dans le cas de certaines mutations de la TTR si effectuée précocement dans l'évolution de la maladie sans atteinte cardiaque. La transplantation plus tard au cours de la maladie conduit souvent à une cardiomyopathie et à une neuropathie amyloïde évolutive due à un mauvais repliement et au dépôt de protéine TTR de type sauvage sur des dépôts amyloïdes préexistants.
Plusieurs médicaments se sont révélés stabiliser les tétramères de TTR circulants (transthyrétine) dans le plasma, ce qui inhibe le mauvais repliement de TTR et la formation de fibrilles et ralentit efficacement la progression de la maladie neurologique, tout en préservant la qualité de vie. Ces stabilisants du TTR (transthyrétine) comprennent le diflunisal, un anti-inflammatoire générique qui est largement disponible et le tafamidis (4, 5).
Le silençage génique de la TTR (transthyrétine) par un ARN anti-sens ou un ARN interférant pour bloquer la traduction du TTR ARNm en protéine réduit efficacement les taux de TTR sériques, améliore l'évolution neurologique chez environ 50% des patients et semble en mesure de réparer les nerfs lésés chez certains patients (6, 7). Le patisiran, l'inotersen et le vutrisiranun médicaments silençant les gènes, sont disponibles.
Un essai du vutrisiran, un silençant de gène de deuxième génération, a démontré de meilleurs résultats fonctionnels chez les patients atteints de polynévrite amyloïde familiale (8). Les données préliminaires d'un autre essai suggèrent que les gene silenceurs peuvent être efficaces dans le traitement de la cardiomyopathie chez les patients atteints d'amylose ATTR (9).
ATTRwt (wild-type ATTR; précédemment appelée amyloïdose systémique sénile ou SSA [senile systemic amyloidosis])
Dans l'ATTRwt (wild-type ATTR; précédemment appelée amyloïdose systémique sénile ou SSA [senile systemic amyloidosis]):
Médicaments stabilisants du tétramer
L a stabilisation de la TTR par le tafamidis chez les patients atteints de cardiomyopathie amyloïde ATTR ou ATTRwt a permis de réduire la mortalité toutes causes confondues et les hospitalisations pour cause cardiovasculaire (5). Des essais cliniques sont en cours pour examiner l'effet des silenceurs du gène TTR sur la cardiomyopathie qui se produit chez les patients qui ont une amylose ATTRwt ainsi que sur la cardiomyopathie des patients qui ont une amylose ATTR caractérisée par une protéine mutante (10).
Contrairement à l’amylose héréditaire ATTR, la transplantation hépatique n’est pas efficace chez les patients atteints d’ATTRwt car la protéine amyloïdogène est une TTR structurellement normale.
Amylose AA (amyloïde A)
Dans l'amylose AA provoquée par la fièvre méditerranéenne familiale, la colchicine orale est efficace.
Pour les autres types d'AA (amyloïde A), le traitement est dirigé contre l'infection, la maladie inflammatoire, ou le cancer sous-jacents.
La colchicine ou des médicaments anti-IL1, anti-IL6 ou anti-TNF peuvent être utilisés pour interrompre la signalisation des cytokines, diminuant ainsi le processus inflammatoire menant à la production hépatique d'amyloïde A sérique.
Références pour le traitement
1. Sanchorawala V, Sun F, Quillen K, et al: Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem cell transplantation: 20-year experience. Blood 126: 2345–2347, 2015. doi: 10.1182/blood-2015-08-662726
2. Kastritis E, Palladini G, Minnema MC, et al: Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. N Engl J Med 385(1):46-58, 2021. doi:10.1056/NEJMoa2028631
3. Quarta CC, Fontana M, Damy T, et al: Changing paradigm in the treatment of amyloidosis: From disease-modifying drugs to anti-fibril therapy. Front Cardiovasc Med 9:1073503, 2022. doi:10.3389/fcvm.2022.1073503
4. Berk JL, Suhr OB, Obici L, et al: Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 310: 2658–2667, 2013. doi: 10.1001/jama.2013.283815
5. Maurer MS, Schwartz JH, Gundapaneni B, et al: Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 379:1007–1016, 2018.
6. Adams D, Gonzalez-Duarte A, O'Riordan WD, et al: Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 379:11–21, 2018.
7. Benson MD, Waddington-Cruz M, Berk JL, et al: Inotersen treatment for patients with transthyretin amyloidosis. N Engl J Med 379:22–31, 2018.
8. Adams D, Tournev IL, Taylor MS, et al: Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid 30(1):1-9, 2023. doi:10.1080/13506129.2022.2091985
9. Maurer MS, Fontanta MA, Berk JL, et al: Primary results from APOLLO-B, a phase 3 study of patisiran in patients with transthyretin-mediated amyloidosis with cardiomyopathy. Abstract presented at International Symposium of Amyloidosis, September 2022, Heidelberg Germany.
10. Writing Committee, Kittleson MM, Ruberg FL, et al: 2023 ACC Expert Consensus Decision Pathway on Comprehensive Multidisciplinary Care for the Patient With Cardiac Amyloidosis: A Report of the American College of Cardiology Solution Set Oversight Committee [published correction appears in J Am Coll Cardiol 81(11):1135, 2023]. J Am Coll Cardiol 81(11):1076-1126, 2023. doi:10.1016/j.jacc.2022.11.022
Pronostic de l'amylose
Le pronostic dépend du type d'amylose et du système d'organes atteint, mais grâce à des soins de support appropriés et spécifiques de la maladie, de nombreux patients ont un excellent pronostic d'espérance de vie.
L'AL compliquée par une cardiomyopathie sévère a le pronostic le moins favorable, avec une survie médiane < 1 an. Non traitée l'amylose provoquée par la transthyrétine (ATTR) progresse habituellement vers le stade cardiaque ou une maladie neurologique en 5 à 15 ans. L'ATTRwt (wild-type ATTR; précédemment appelée amyloïdose systémique sénile ou SSA [senile systemic amyloidosis]) était supposée être généralement à évolution plus lente que toute autre amylose systémique impliquant le cœur; cependant, certains patients qui ont une ATTRwt progressent vers l'insuffisance cardiaque symptomatique et la mort dans un délai médian de 4 ans à compter du diagnostic de biopsie.
Le pronostic de l'amylose AA (amyloïde A) dépend largement de l'efficacité du traitement de la maladie infectieuse, inflammatoire ou maligne sous-jacente.
Points clés
L'amylose est un groupe de maladies dans lesquelles certaines protéines mal repliées s'agrègent en fibrilles insolubles qui se déposent à l'intérieur des organes, ce qui provoque un dysfonctionnement.
De nombreuses protéines différentes sont sujettes à ce mauvais repliement; certaines de ces protéines sont produites du fait d'un défaut génétique ou de certains états pathologiques, tandis que d'autres impliquent des chaînes légères d'immunoglobulines produites par des plasmocytes monoclonaux ou d'autres troubles lymphoprolifératifs cellulaires B.
La protéine amyloïdogène détermine le type de substance amyloïde et l'évolution clinique de la maladie, bien que les manifestations cliniques des différents types puissent se chevaucher.
De nombreux organes peuvent être touchés, mais l'atteinte cardiaque a un pronostic particulièrement défavorable; la cardiomyopathie amyloïde conduit généralement à une dysfonction diastolique, à l'insuffisance cardiaque, et au bloc cardiaque ou à l'arythmie.
Le diagnostic est établi par la biopsie; le type d'amyloïdose est déterminé par divers tests immunologiques, génétiques et biochimiques. La spectrométrie de masse est la méthode la plus sensible et la plus spécifique de typage des amyloïdes.
Des soins de support appropriés permettront de soulager les symptômes et d'améliorer la qualité de la vie; la transplantation d'organe peut aider des patients sélectionnés.
Traiter le processus sous-jacent; dans l'amylose AL (light chain) due à des troubles des plasmocytes ou lymphoprolifératifs, la chimiothérapie peut être très efficace; dans l'amylose AA (amyloïde A) secondaire, les thérapies anti-infectieuses et anti-inflammatoires peuvent être utiles.
Dans l'amylose héréditaire ATTR, les petites molécules thérapeutiques stabilisatrices et les médicaments qui rendent silencieux les gènes bloquent ou même peuvent renverser la détérioration neurologique; en cas de cardiomyopathie amyloïde (ATTR ou ATTRwt), le tafamidis diminue la mortalité de toutes causes confondues et les hospitalisations pour cause cardiovasculaire.







