


La pemphigoïde bulleuse est une dermatose bulleuse auto-immune provoquant des lésions bulleuses prurigineuses généralisées chez le patient âgé. L'atteinte de la muqueuse est rare. Le diagnostic repose sur la biopsie cutanée et l'immunofluorescence de la peau et du sérum. Les corticostéroïdes topiques ou systémiques sont utilisés initialement. La plupart des patients doivent suivre un traitement d'entretien à long terme, pour lequel toute une variété d'immunosuppresseurs peut être utilisée.
Une lésion bulleuse correspond à une collection liquidienne ≥ 10 mm de diamètre.
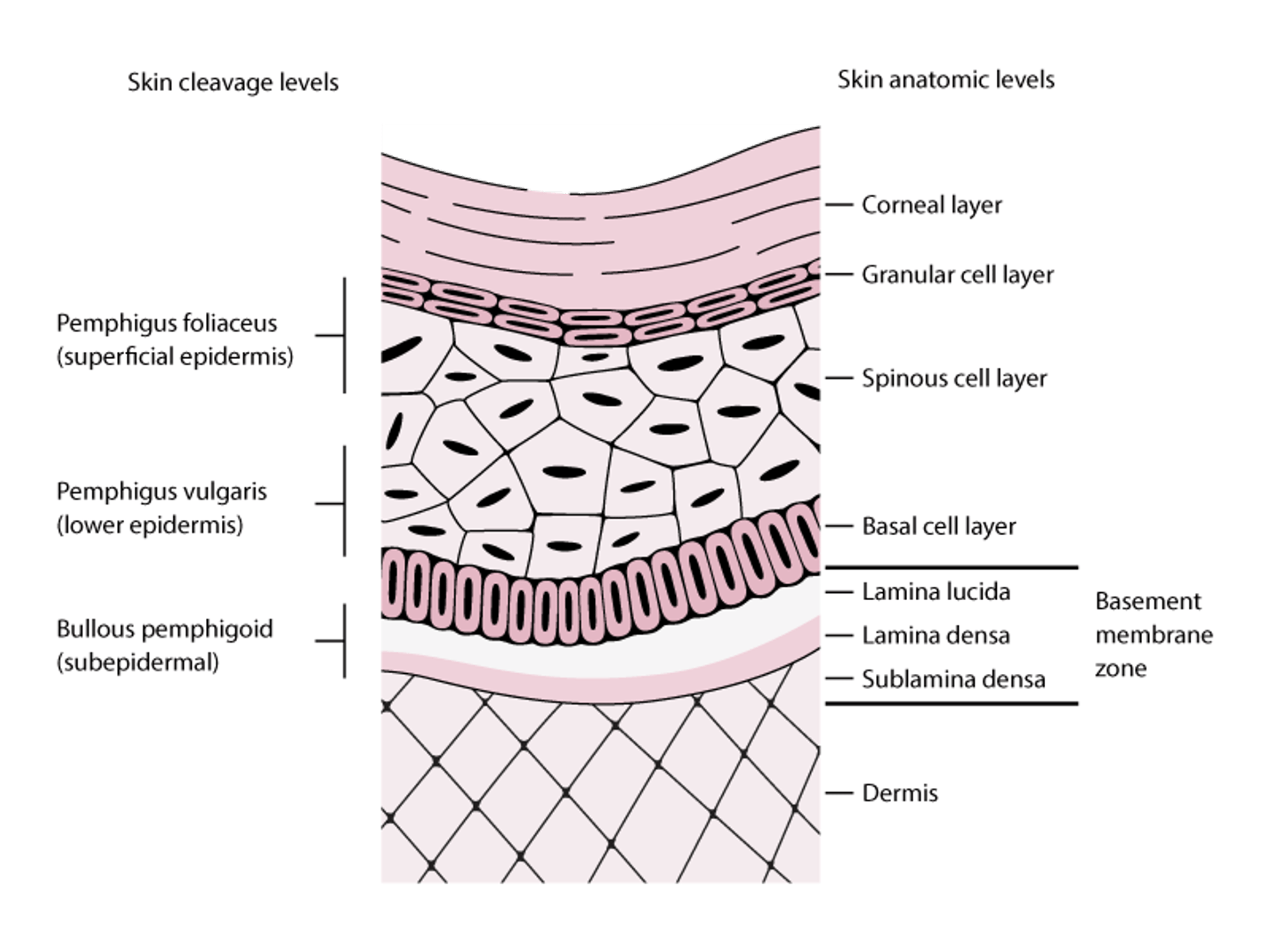
La pemphigoïde bulleuse est principalement observée chez les patients de > 60 ans, mais peut survenir chez les enfants. Les auto-anticorps IgG se lient à certains antigènes hémidesmosomiaux (BPAg1 [BP230], BPAg2 [BP180]), ce qui entraîne l'activation du complément pour former une papule sous-épidermique (voir figure Niveaux de clivage cutané dans le pemphigus et l'épidermolyse bulleuse).
Niveaux de clivage cutané dans le pemphigus et l'épidermolyse bulleuse
Les bulles de pemphigus foliacé se forment dans les couches superficielles de l'épiderme. Les bulles de pemphigus vulgaris peuvent se former à n'importe quel niveau de l'épiderme, mais se forment généralement dans les parties inférieures de l'épiderme. Des bulles de pemphigoïde bulleuse se forment sous l'épiderme (lamina lucida de la zone de la membrane basale). Dans cette figure, la région de la membrane basale est agrandie de manière non proportionnelle pour mettre en évidence ses couches. |

Étiologie de la pemphigoïde bulleuse
Aucune cause de la pemphigoïde bulleuse n'a été prouvée; cependant, les déclencheurs suivants ont été suggérés:
Médicaments (dont furosémide, spironolactone, l'oméprazole, anticorps monoclonaux contre PD-1 et PD-L1 [p. ex., durvalumab, nivolumab, pembrolizumab], sulfasalazine, pénicilline, pénicillamine, étanercept, antipsychotiques, inhibiteurs de la dipeptidyl peptidase-4)
Déclencheurs physiques (dont les traumatismes, la radiothérapie pour le cancer du sein, le rayonnement UV et l'anthraline)
Les affections cutanées (y compris le psoriasis, le lichen plan et certaines infections)
Troubles (diabète sucré, polyarthrite rhumatoïde, rectocolite ulcéro-hémorragique et sclérose en plaques)
Les facteurs génétiques et environnementaux peuvent jouer un rôle.
Les déclencheurs peuvent induire une réaction auto-immune en mimant les séquences moléculaires dans la membrane basale épidermique (mimétisme moléculaire, comme c'est le cas avec les médicaments et, éventuellement, les infections), en exposant ou en modifiant les antigènes de l'hôte normalement tolérés (comme avec les déclencheurs physiques et certaines maladies), ou par d'autres mécanismes. L'apparition de nouveaux épitopes correspond au recrutement de lymphocytes autoréactifs contre des antigènes de l'hôte normalement tolérés, ce qui contribue à la chronicité et à l'évolution de la maladie.
Certains troubles du système nerveux central et psychiatriques peuvent précéder la pemphigoïde bulleuse, en particulier la sclérose en plaques et la schizophrénie, mais également des démences, des hémorragies intracrâniennes, des accidents vasculaires cérébraux, des troubles délirants et de la personnalité et la maladie de Parkinson. Dans une moindre mesure, ces troubles peuvent être précédés d'une pemphigoïde bulleuse. Les causes communes hypothétiques comprennent une réponse immunitaire croisée entre les antigènes neuronaux et cutanés (BPAg1 est exprimé dans le système nerveux central), ainsi que le déclenchement par certains médicaments utilisés pour traiter les troubles (p. ex., antipsychotiques phénothiazine, spironolactone); cependant, un mécanisme de déclenchement par des médicaments n'est pas compris.
Symptomatologie de la pemphigoïde bulleuse
Le prurit est le premier symptôme de la pemphigoïde bulleuse. Les lésions cutanées peuvent ne pas se développer avant plusieurs années. Souvent, des bulles tendues caractéristiques se développent sur la peau du tronc et dans les zones de flexion et intertrigineuses. Les bulles peuvent se développer sur une peau d'aspect normal ou être précédées de plaques érythémateuses ou urticariennes. La maladie localisée peut survenir au niveau des sites des traumatismes, des stomies, de la région anogénitale et de la partie basse des jambes. La pemphigoïde dyshidrotique est une forme rare de pemphigoïde bulleuse qui affecte les mains et les pieds et peut ressembler à une dermatite dyshidrotique (une forme de dermatite des mains et des pieds) au nievau des paumes. Généralement les bulles ne se rompent pas, mais si tel est le cas, la guérison est rapide.
Des lésions polymorphiques, à disposition annulaire, érythémateuses, œdémateuses, urticariennes, à disposition parfois annulaires avec vésicules ou non en périphérie peuvent apparaître. Rarement, de petites vésicules se développent sur la muqueuse. La leucocytose et une éosinophilie sont fréquentes, mais la fièvre est rare. Le signe de Nikolsky, dans lequel les couches supérieures de l'épiderme se déplacent latéralement par légère pression ou frottement de la peau adjacente à une bulle, est négatif.
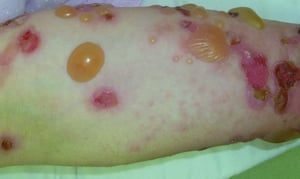
© Springer Science+Business Media
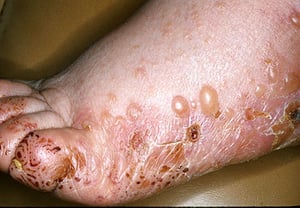
Image provided by Thomas Habif, MD.
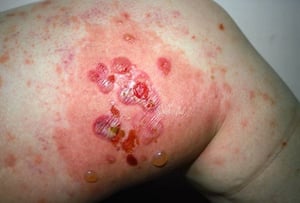
Image courtesy of Karen McKoy, MD.
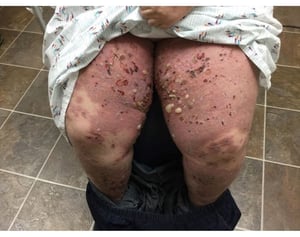
Image courtesy of Daniel M. Peraza, MD.

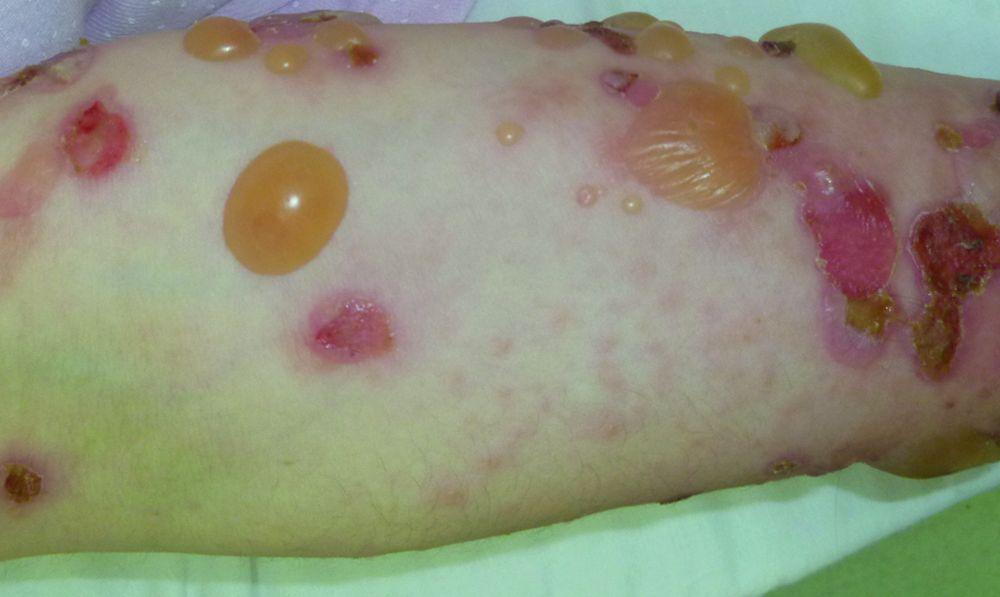
© Springer Science+Business Media

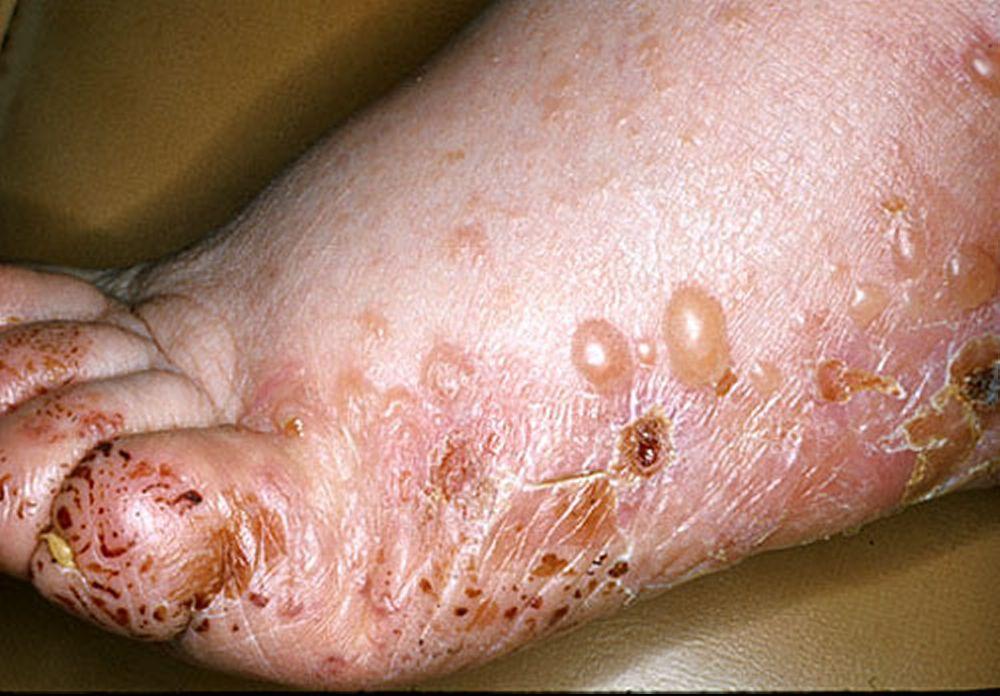
Image provided by Thomas Habif, MD.

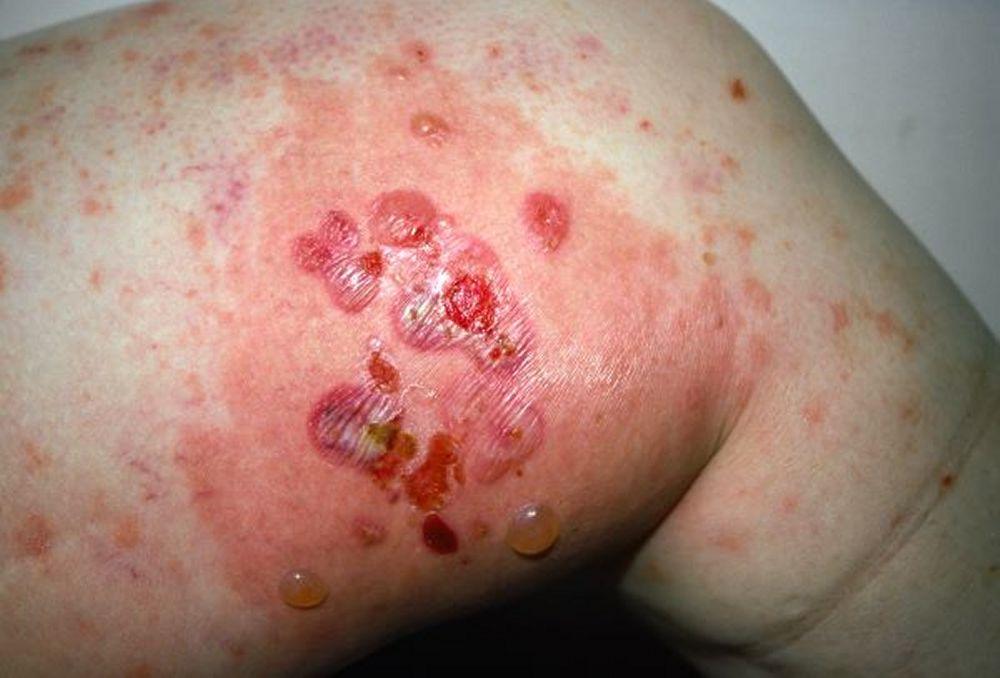
Image courtesy of Karen McKoy, MD.

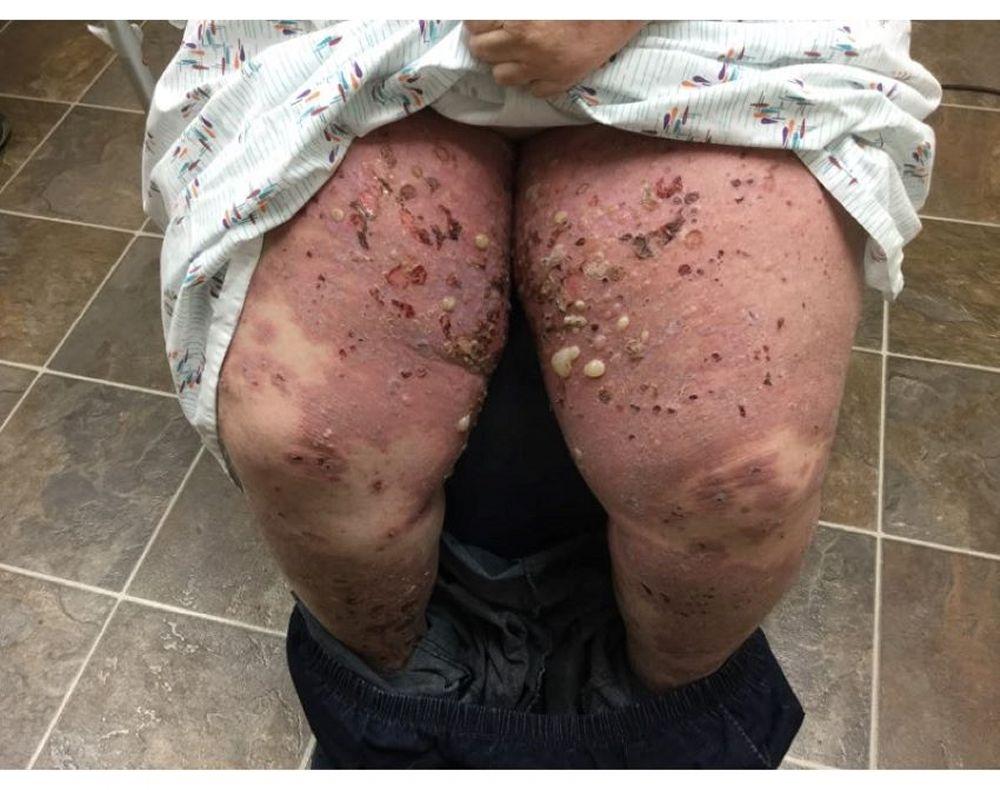
Image courtesy of Daniel M. Peraza, MD.
Diagnostic de la pemphigoïde bulleuse
Biopsie cutanée et titres d'IgG
Si des bulles se développent, la pemphigoïde doit être différenciée du pemphigus vulgaire, un trouble de mauvais pronostic; la différenciation est généralement possible en utilisant des critères cliniques ( see table Différences entre la pemphigoïde bulleuse et le pemphigus vulgaire).
Les tests permettent de différencier la pemphigoïde bulleuse du pemphigus vulgaire, de la maladie à IgA linéaires, de l'érythème polymorphe, des éruptions médicamenteuses, de la pemphigoïde des muqueuses, de la pemphigoïde paranéoplasique, de la dermatite herpétiforme et de l'épidermolyse bulleuse acquise.
En cas de suspicion de pemphigoïde bulleuse, une biopsie cutanée est effectuée pour rechercher l'histologie, ainsi qu'un test d'immunofluorescence directe. Les prélèvements provenant de et d'autour de la lésion elle-même sont souvent utilisés pour l'histologie, mais des prélèvements de peau non atteinte (souvent environ à 3 mm du bord d'une lésion) sont utilisés pour diriger l'immunofluorescence directe. La bulle de la pemphigoïde est sous-épidermique, contenant souvent de nombreux neutrophiles et éosinophiles. L'immunofluorescence directe montre des dépôts linéaires d'IgG et de complément le long de la membrane basale (jonction dermo-épidermique). L'immunofluorescence indirecte montre des dépôts d'IgG circulants sur la face épidermique d'une préparation séparée par le sel d'une peau normale (c'est-à-dire, substrat de test).
Les anticorps IgG contre BPAG1 et BPAg2 sont dosés par méthode immuno-enzymatique (ELISA). Des anticorps IgG circulants sont présents chez environ 3/4 des patients.
Pronostic de la pemphigoïde bulleuse
La pemphigoïde bulleuse est une maladie chronique, potentiellement mortelle, en particulier sans traitement. Bien que les thérapies topiques et systémiques soient utiles, elles peuvent avoir des effets indésirables.
La rémission est typique en quelques mois, mais le traitement est parfois nécessaire pendant plusieurs années.
Traitement de la pemphigoïde bulleuse
Corticostéroïdes, topiques ou par voie orale
Médicaments anti-inflammatoires
Médicaments immunosuppresseurs ou biologiques
Des corticostéroïdes topiques de forte puissance (p. ex., la crème de clobétasol à 0,05%) doivent être utilisés dans la maladie localisée et peuvent réduire la dose requise de médicaments systémiques.
En cas d'atteinte généralisée, un traitement systémique avec la prednisone 60 à 80 mg par voie orale 1 fois/jour est souvent nécessaire, cette posologie peut ensuite être abaissée jusqu'à une dose d'entretien ≤ 10 à 20 mg/jour après plusieurs semaines. La plupart des patients ont une rémission après 2 à 10 mois, mais le traitement peut devoir être poursuivi pendant plusieurs années avant que le processus pathologique s'éteigne suffisamment pour permettre l'arrêt du traitement. Si un traitement à long terme est nécessaire, une nouvelle bulle toutes les quelques semaines ne nécessite pas d'augmenter la dose de prednisone.
La pemphigoïde bulleuse peut parfois être contrôlée par l'activité anti-inflammatoire de certains médicaments tels que l'association de tétracycline ou de minocycline et de nicotinamide. D'autres options de traitement comprennent la monothérapie par la dapsone, la sulfapyridine, ou l'érythromycine. Les IgIV ont été efficaces dans certaines formes sévères.
En cas de maladie généralisée et résistante, et parfois afin de diminuer la dose de corticostéroïdes dans la maladie chronique, les immunosuppresseurs tels que le méthotrexate, l'azathioprine, le cyclophosphamide, le mycophénolate mofétil et la cyclosporine peuvent être utilisés. Parmi les agents biologiques, le rituximab et l'omalizumab peuvent être utilisés.
Points clés
La pemphigoïde affecte généralement les patients > 60 ans et est auto-immune et idiopathique.
Le prurit peut précéder de plusieurs années le développement d'une éruption, et l'atteinte de la muqueuse est rare.
Biopsie cutanée pour histologie et l'immunofluorescence et mesure les auto-anticorps circulants.
Traiter les patients par des corticostéroïdes topiques de haute puissance si possible pour éviter ou minimiser l'utilisation de corticostéroïdes systémiques.
Un traitement anti-inflammatoire, immunosuppresseur et biologique peut être utilisé pour diminuer la dose de corticostéroïdes.
Les symptômes diminuent généralement en quelques mois, mais un traitement est parfois nécessaire pendant plusieurs années.







