


La myosite auto-immune est caractérisées par des anomalies inflammatoires et dégénératives soit uniquement musculaires (polymyosite, myopathie nécrosante à médiation immunitaire), soit à la fois cutanées et musculaires (dermatomyosite). Les manifestations comprennent une faiblesse musculaire symétrique, une douleur occasionnelle et, une fibrose de remplacement du tissu musculaire, parfois avec une atrophie, principalement des groupes de muscles proximaux des membres. Le diagnostic repose sur des signes cliniques et des examens pratiqués sur les muscles, qui peuvent comprendre les taux de créatine kinase, l'IRM, l'électromyographie et la biopsie musculaire. Plusieurs types de myosite donnent des manifestations pulmonaires et cardiaques. Le traitement repose sur les corticostéroïdes, associés à des immunosuppresseurs et/ou à des IgIV.
La myosite auto-immune est plus fréquente chez les femmes que chez les hommes dans un rapport de 2:1. L'incidence est 3 à 4 fois plus élevée chez les Noirs que chez les Blancs. Ces troubles peuvent apparaître à tout âge, mais surviennent le plus souvent entre 40 et 60 ans ou, chez l'enfant, entre 5 et 15 ans.
Étiologie de la myosite auto-immune
La cause de la myosite auto-immune semble être une réaction auto-immune dirigée contre le tissu musculaire chez des individus génétiquement prédisposés. Des clusters familiaux existent et des sous-types d'antigène leucocytaire humain (HLA) sont associés à la myosite. Par exemple, les allèles de l'haplotype héréditaire 8.1 (HLA-DRB1*03-DQA1*05-DQB1*02) augmentent le risque de polymyosite, de dermatomyosite et de maladie pulmonaire interstitielle. Des événements déclenchants potentiels peuvent être une myosite virale et une affection maligne sous-jacente. L'association tumeur maligne dermatomyosite (moins en cas de polymyosite) suggère qu'un cancer peut entraîner une myosite par suite d'une réaction auto-immune contre un antigène commun au muscle et à la tumeur.
Physiopathologie de la myosite auto-immune
Les modifications anatomopathologiques comprennent des lésions cellulaires et atrophies, avec des degrés variables d'inflammation. Les muscles des mains, des pieds et du visage sont moins atteints que d'autres muscles squelettiques. L'atteinte des muscles dans le pharynx et la partie supérieure de l'œsophage et parfois dans le cœur peut altérer les fonctions de ces organes. L'inflammation peut toucher les articulations et les poumons, en particulier en présence d'anticorps anti-synthétase.
La dermatomyosite est caractérisée par des dépôts d'immuns-complexes dans les vaisseaux et est considérée comme une vasculopathie médiée par le complément. En revanche, la polymyosite est caractérisée par une lésion musculaire directe à médiation par les lymphocytes T et les myopathies nécrosantes à médiation immunitaire sont caractérisées par des infiltrats à prédominance de macrophages et une myophagocytose.
Classification de la myosite auto-immune
La myosite auto-immune peut être classée en 4 groupes, qui sont principalement basés sur l'histopathologie et la présentation clinique:
Polymyosite
Dermatomyosite
Myopathies nécrosante à médiation immunitaire
Myosite à corps d'inclusion
La dermatomyosite peut être distinguée de la polymyosite par les signes cutanés caractéristiques de la dermatomyosite (voir Symptomatologie). L'histopathologie musculaire diffère également. La dermatomyosite et la polymyosite peuvent se manifester sous la forme de maladies musculaires pures ou faire partie d'un syndrome antisynthétase qui peut être associé à une arthrite (habituellement non érosive), à de la fièvre, à une maladie pulmonaire interstitielle, à une hyperkératose de la face radiale des doigts (mains du mécanicien) et à un syndrome de Raynaud.
Les myopathies nécrosantes à médiation immunitaire comprennent le plus souvent la "signal recognition particle (SRP) antibody–related myositis" et la myosite induite par les statines; elles ont généralement une présentation agressive, avec des taux de créatine kinase (CK) très élevés et n'impliquent pas d'organes extramusculaires (1).
La myosite à inclusion entraîne une faiblesse musculaire proximale des jambes, mais touche souvent des muscles distaux (p. ex., les muscles des mains et des pieds) et est souvent associée à une atrophie musculaire. Elle se développe à un âge avancé, a une progression plus lente et ne répond généralement pas au traitement immunosuppresseur.
La myosite auto-immune peut également s'associer à d'autres troubles rhumatismaux auto-immuns, p. ex., le lupus érythémateux disséminé, la sclérose systémique, la connectivite mixte. Ces patients présentent la symptomatologie des troubles associés en plus de ceux de la myosite (qui se manifestent soit sous forme de dermatomyosite soit de polymyosite).
Référence pour la classification
1. Lundberg IE, Fujimoto M, Vencovsky J, et al. Idiopathic inflammatory myopathies. Nat Rev Dis Primers 7(1):86, 2021. doi:10.1038/s41572-021-00321-x
Symptomatologie de la myosite auto-immune
L'apparition de la myosite auto-immune peut être aiguë (en particulier chez l'enfant) ou insidieuse (surtout chez l'adulte). Peuvent également survenir des polyarthralgies, un syndrome de Raynaud, une dysphagie, des symptômes pulmonaires (p. ex., toux, dyspnée) et des symptômes généraux (en particulier fièvre, asthénie et perte de poids). La maladie grave est caractérisée par une dysphagie, une dysphonie et/ou une faiblesse diaphragmatique.
Cette faiblesse musculaire peut évoluer en quelques semaines ou quelques mois. Cependant, il faut la destruction de 50% des fibres musculaires pour entraîner une faiblesse musculaire symptomatique (c'est-à-dire, une faiblesse musculaire qui indique une myosite avancée). Le patient a du mal à lever les bras au-dessus de la tête, à monter des marches ou à se lever de son siège. Parfois, une sensibilité et une atrophie musculaires se développent. Certains patients sont confinés dans un fauteuil roulant ou au lit, du fait de la faiblesse des ceintures scapulaire et pelvienne. Les fléchisseurs du cou peuvent être sévèrement affectés et le malade a du mal à soulever la tête de l'oreiller. L'atteinte des muscles pharyngés et œsophagiens supérieurs peut altérer la déglutition et prédisposer à une fausse route. Les muscles des mains, des pieds et du visage ne sont pas touchés sauf dans la myosite à corps d'inclusion, dans laquelle l'atteinte distale, en particulier des mains, est caractéristique. Des rétractions des membres se développent rarement.
Les manifestations articulaires comprennent une polyarthralgie ou une polyarthrite, associées à un gonflement et à d'autres caractéristiques de l'arthrite non déformante. Elles apparaissent plus souvent dans le sous-groupe de patients qui ont des anticorps anti-Jo-1 ou d'autres anticorps anti-synthétase.
Les atteintes viscérales (sauf celle de l'œsophage du pharynx et supérieure) sont moins fréquentes dans la myosite auto-immune que dans certains autres troubles rhumatismaux (p. ex., lupus érythémateux disséminé, sclérodermie). Parfois, et en particulier en présence d'anticorps anti-synthétase, une maladie pulmonaire interstitielle (qui se manifeste par une toux et une dyspnée) domine le tableau. Une atteinte cardiaque, en particulier des troubles de la conduction et un dysfonctionnement ventriculaire, peuvent survenir. Les symptômes digestifs, plus fréquents chez l'enfant, sont provoqués par une vascularite associée et peuvent comprendre une douleur abdominale, une hématémèse, un méléna et une perforation intestinale ischémique.
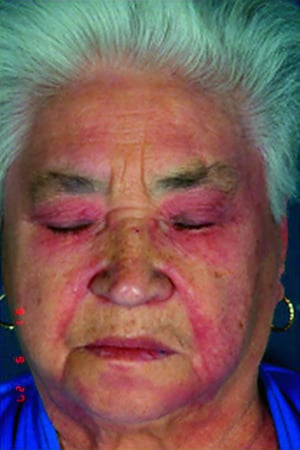
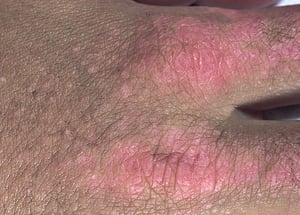
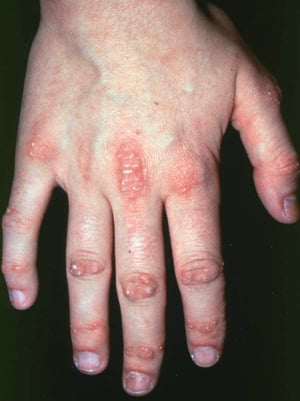
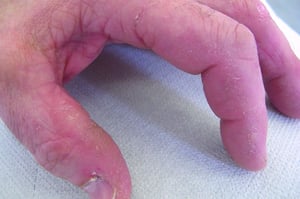
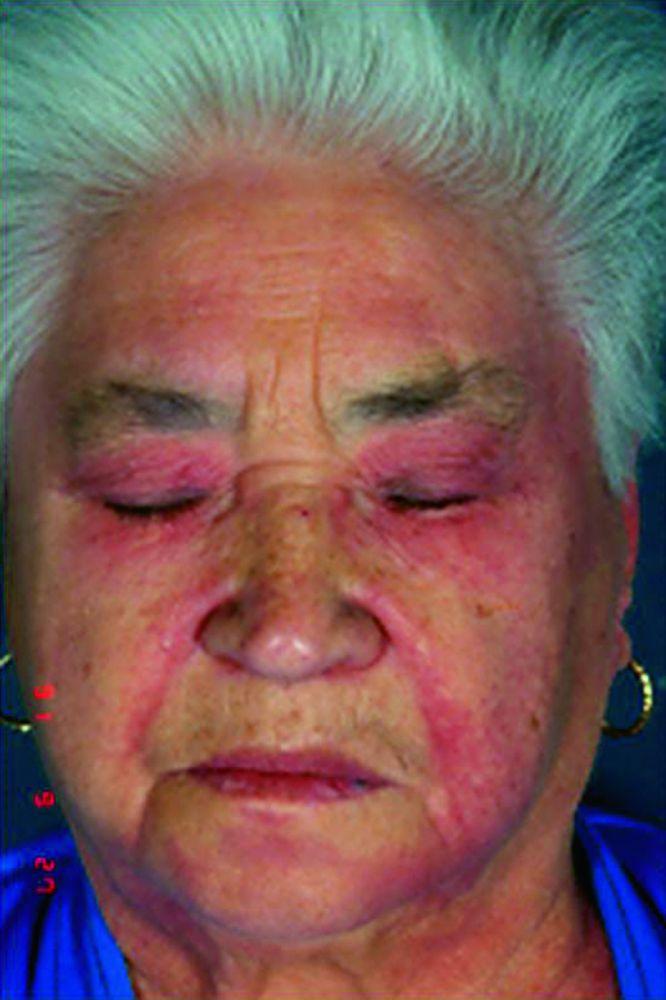
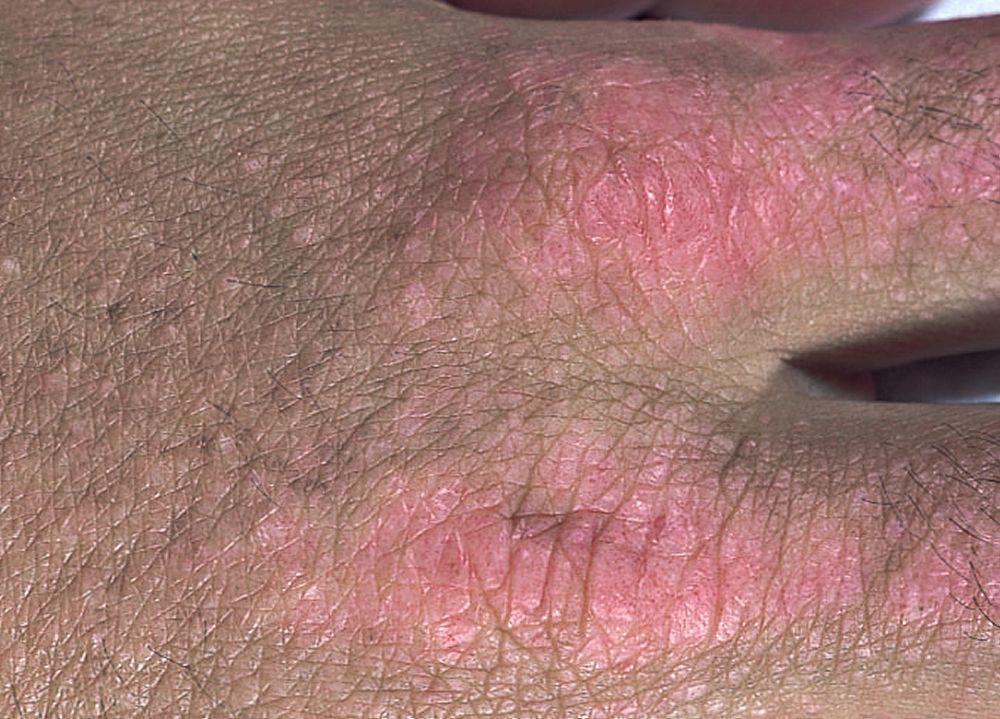
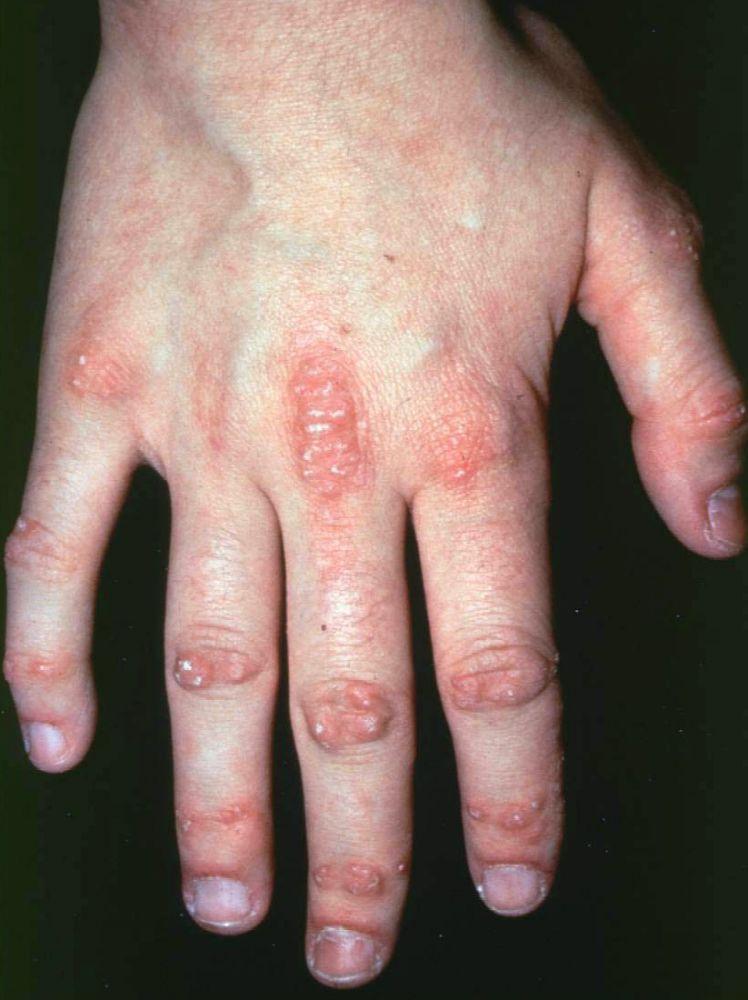
Les atteintes cutanées, qui surviennent dans la dermatomyosite, sont érythémateuses ou violines. Une photosensibilité et une ulcération de la peau sont visibles. L'œdème périorbitaire avec aspect violacé (érythème héliotrope) est relativement spécifique à la dermatomyosite. Ailleurs, l'éruption peut être légèrement surélevée et lisse ou squameuse; elle peut apparaître sur le front, le V du cou et les épaules, le thorax et le dos, les avant-bras et le bas des jambes, la partie latérales des cuisses, les coudes et les genoux, les malléoles médiales et les faces dorsales des articulations interphalangiennes et métacarpophalangiennes proximales (les papules de Gottron, un marqueur relativement spécifique). La base et les côtés des ongles peuvent être épaissis ou hyperhémiques. Une dermite desquamante associée à des fissures de la peau peut être observée sur la surface radiale des doigts. Des calcifications sous-cutanées et musculaires peuvent survenir, en particulier chez l'enfant. Les lésions cutanées primitives guérissent généralement sans laisser de traces, mais des signes secondaires peuvent apparaître (p. ex., pigmentation brunâtre, atrophie, néovascularisation persistante, sclérose). L'éruption sur le cuir chevelu peut sembler psoriasiforme et être intensément prurigineuse.
Des anomalies cutanées caractéristiques peuvent survenir en l'absence de maladie musculaire, auquel cas la maladie est appelée dermatomyosite amyopathique.
© Springer Science+Business Media
© Springer Science+Business Media
© Springer Science+Business Media
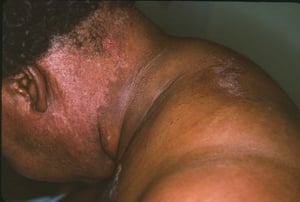
Image courtesy of Karen McKoy, MD.
© Springer Science+Business Media

© Springer Science+Business Media

© Springer Science+Business Media

© Springer Science+Business Media

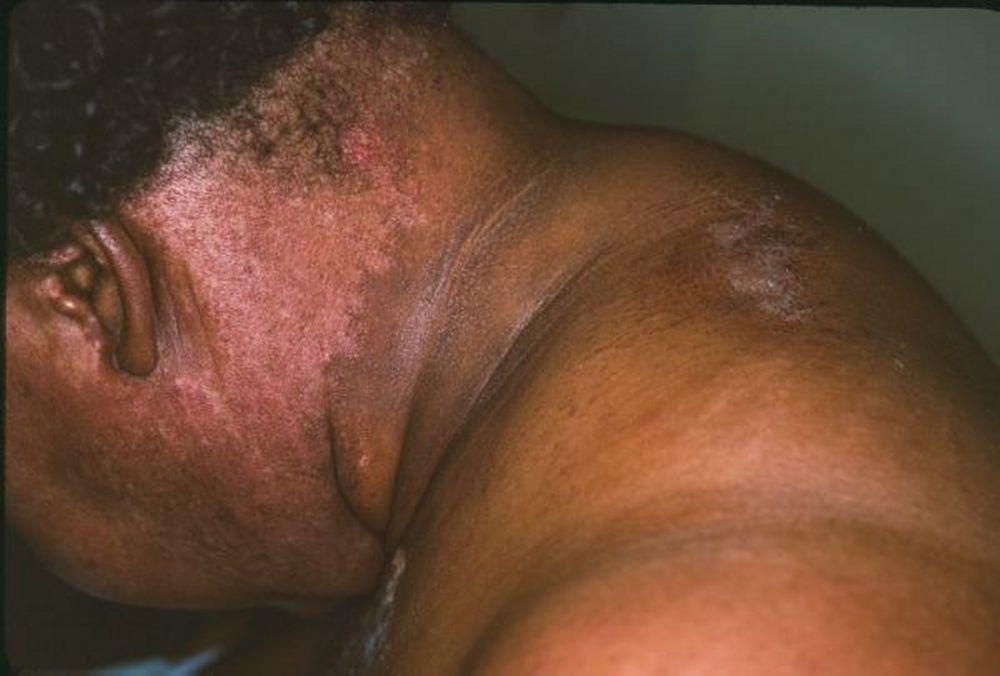
Image courtesy of Karen McKoy, MD.

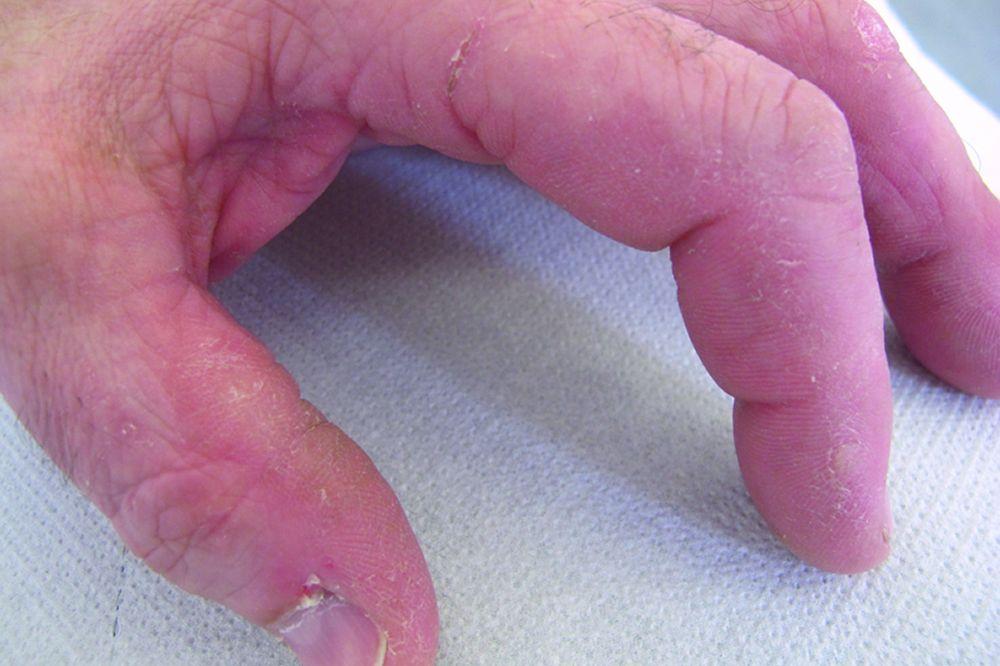
© Springer Science+Business Media
Diagnostic de la myosite auto-immune
Critères cliniques
Biopsie musculaire (définitif)
La myosite auto-immune doit être suspectée en cas de faiblesse musculaire proximale avec ou sans sensibilité musculaire. Une dermatomyosite doit être suspectée devant des signes de myosite et de signes cutanés compatibles avec une dermatomyosite. L'établissement du diagnostic de myosite auto-immune exige le plus possible des 5 critères suivants:
Faiblesse des muscles proximaux
Éruption caractéristique
Taux sériques d'enzymes musculaires élevés (si la creatine kinase [CK] n'est pas élevée, aminotransférases ou aldolase [qui sont beaucoup moins spécifiques que les CK])
Anomalies caractéristiques électromyographiques ou IRM musculaire
Modifications décelées à la biopsie musculaire (diagnostic de certitude)
Les signes à la biopsie peuvent varier, mais une inflammation chronique avec dégénérescence musculaire et une certaine régénération est typique. La polymyosite et la dermatomyosite peuvent souvent être distinguées par une biopsie musculaire. Un diagnostic de certitude apporté par la biopsie musculaire est recommandé avant le traitement de la polymyosite pour exclure d'autres troubles musculaires, tels que ceux dus aux enzymes manquantes ou défectueuses, à une myosite nécrosante et à la rhabdomyolyse postvirale. La biopsie musculaire n'est généralement pas nécessaire lorsque les signes cutanés sont caractéristiques de la dermatomyosite. Il n'y a pas de signe cutané pathognomique de dermatomyosite à la biopsie, mais l'absence d'immunofluorescence directe permet de distinguer l'éruption de l'éruption chez les patients atteints de lupus érythémateux disséminé.
Pour augmenter la sensibilité des résultats de la biopsie, le prélèvement de biopsie doit être obtenu à partir d'un muscle qui a l'une ou plusieurs des caractéristiques suivantes:
Faiblesse à l'examen clinique
Œdème musculaire identifié sur l'IRM
Paire controlatérale d'un muscle, anormale à l'électromyographie
Des examens de laboratoire peuvent augmenter ou diminuer la suspicion de la maladie, évaluer sa gravité, identifier les chevauchements et détecter les complications. Les auto-anticorps doivent être testés. Les anticorps antinucléaires (AAN) sont positifs jusque dans 80% des cas de dermatomyosite et de polymyosite. Si le test AAN est positif, d'autres tests pour des types spécifiques d'anticorps sont importants pour augmenter la suspicion de syndrome de chevauchement.
L'évolution et les manifestations cliniques sont associées à des anticorps particuliers tels que décrits dans le tableau Autoanticorps dans la myosite auto-immune. Les relations entre ces auto-anticorps et la physiopathologie de la maladie sont mal comprises, bien que les anticorps anti-Jo-1 représentent un marqueur intéressant de l'alvéolite fibrosante, de la fibrose pulmonaire, de l'arthrite et du syndrome de Raynaud. Il n'y a pas d'anticorps spécifiques de la polymyosite.
Les preuves d'un risque accru de cancer sont relativement solides dans la dermatomyosite et moins dans la polymyosite. Par conséquent, une recherche de cancer doit être envisagée chez tout patient de ≥ 40 ans qui a une dermatomyosite ou chez les patients de ≥ 60 ans qui ont une polymyosite parce que ces patients ont souvent des cancers non diagnostiqués. Le dépistage doit comprendre au moins un examen clinique qui comprend l'examen des seins, du pelvis et du rectum (avec recherche d'hémorragie occulte); NFS; profil biochimique; mammographie; analyse d'urine; rx thorax; et tous autres tests appropriés basés en fonction de l'âge du patient.
Les investigations supplémentaires doivent être basées sur l'anamnèse et les signes d'examen clinique. Certains experts conseillent une TDM du thorax et abdomino-pelvienne ainsi que la coloscopie, en particulier chez les patients qui ont une dermatomyosite. Les patients jeunes sans symptômes de cancer ne doivent pas subir de dépistage.
Pronostic de la myosite auto-immune
De longues rémissions (même une guérison apparente) surviennent dans une proportion allant jusqu'à 50% des patients traités, dans les 5 ans, en général chez l'enfant. Cependant, une rechute peut avoir lieu à tout moment. Le taux de survie globale à 5 ans est de 75% et est plus élevé chez l'enfant.
Chez l'adulte, la mort survient par faiblesse musculaire sévère et progressive, dysphagie, dénutrition, pneumopathies par inhalation ou défaillance respiratoire par infection surajoutée.
La mort chez l'enfant atteint de dermatomyosite peut être le résultat d'une vascularite intestinale.
La dermatomyosite et la polymyosite ont été associées à un risque accru de cancer. Une tumeur maligne, si elle est présente, détermine généralement le pronostic global.
Traitement de la myosite auto-immune
Corticostéroïdes
Des immunosuppresseurs (p. ex., méthotrexate, azathioprine, mycophénolate mofétil, rituximab, tacrolimus)
Immunoglobulines IV
Les activités physiques doivent être légèrement réduites jusqu'à ce que l'inflammation disparaisse.
Les corticostéroïdes s'imposent au début du traitement. Dans une maladie aiguë, les adultes reçoivent de la prednisone 1 mg/kg habituellement de 40 à 60 mg par voie orale 1 fois/jour. Dans le cas d'une maladie grave avec dysphagie ou faiblesse musculaire respiratoire, le traitement débute habituellement par un corticostéroïde à forte dose (p. ex., méthylprednisolone 0,5 à 1 g IV 1 fois/jour pendant 3 à 5 jours).
Les mesures répétées de l'activité de creatine kinase (CK) permettent d'évaluer précocement l'efficacité du traitement. Cependant, en cas d'atrophie musculaire disséminée, les taux sont parfois normaux malgré une myosite chronique active. Les signes IRM d'un œdème musculaire ou des taux élevés de CK permettent de différencier généralement la rechute d'une myosite d'une myopathie induite par les corticostéroïdes. L'aldolase est une alternative, qui est moins spécifique des lésions musculaires que la CK, mais elle peut parfois être positive en cas de myosite et de taux de CK normaux. Lorsque les taux d'enzymes diminuent ou rejoignent la normale chez de nombreux patients en 6 à 12 semaines, suivis plus tard par une amélioration de la force musculaire, la dose de corticostéroïdes peut être progressivement réduite. Si les taux d'enzymes musculaires augmentent à nouveau, la dose de corticostéroïdes est habituellement augmentée en attendant l'effet complet des autres médicaments.
L'objectif global est d'éliminer rapidement l'inflammation tout en minimisant l'exposition aux corticostéroïdes, c'est pourquoi un second médicament (généralement le méthotrexate, le tacrolimus ou l'azathioprine en première intention) est démarré en même temps que les corticostéroïdes ou peu de temps après afin que la prednisone puisse être réduite à une dose maximale de 5 mg/jour dans les 6 mois environ. Les immunoglobulines IV sont une bonne option chez les patients qui ne répondent pas rapidement au traitement, en cas de complications infectieuses dues aux corticostéroïdes à forte dose et à d'autres immunosuppresseurs ou en cas de chimiothérapie. Certains experts peuvent utiliser une association des 3 thérapies dans les cas graves ou en cas de toxicité des corticostéroïdes. Chez l'enfant, il faut prescrire des doses initiales de prednisone de 30 à 60 mg/m2 1 fois/jour.
Parfois, des patients placés de façon chronique sous corticothérapie et à forte dose, présentent une fatigue musculaire croissante après la réponse initiale due à une myopathie indolore cortisonique surajoutée. Chez ces patients, la CK reste normale même si les patients sont plus faibles.
La myosite associée au cancer est plus réfractaire à la corticothérapie. Quand elle est associée à un cancer, la myosite peut disparaître après ablation de la tumeur.
En cas de pathologie auto-immune, le risque d'athérosclérose doit être attentivement suivi. Les patients sous corticostéroïdes au long cours peuvent recevoir un biphosphonate pour prévenir l'ostéoporose. Si un traitement immunosuppresseur d'association est utilisé, les patients doivent recevoir une prophylaxie contre les infections opportunistes, telles que par Pneumocystis jirovecii (voir prévention de la pneumonie à Pneumocystis jirovecii), et des vaccins contre les infections fréquentes (p. ex., pneumonie streptococcique, grippe, COVID-19).
Points clés
La faiblesse musculaire provoquée par la myosite est le plus souvent proximale.
Une éruption héliotropique et des papules Gottron sont spécifiques de la dermatomyosite.
Pour établir le diagnostic, rechercher les caractéristiques éruptions cutanées, faiblesse musculaire, élévation du taux de creatine kinase (CK) et anomalies musculaires à l'électromyographie ou à l'IRM.
Sauf si les patients ont les signes caractéristiques cutanés, effectuer une biopsie musculaire pour confirmer le diagnostic.
Envisager de dépister les patients de ≥ 40 ans ayant une dermatomyosite et les patients de ≥ 60 ans ayant une polymyosite à la recherche d'un cancer.
Traiter les patients par des corticostéroïdes et d'autres immunosuppresseurs.







