


Les gliomes sont des tumeurs primitives qui prennent forme dans le parenchyme cérébral. Les symptômes sont divers et varient selon la localisation, se manifestant par des déficits neurologiques focaux, une encéphalopathie ou des convulsions. Le diagnostic repose principalement sur l'IRM, comprenant à la fois une imagerie pondérée en T1 et T2, de préférence avec rehaussement au gadolinium, suivie d'une biopsie avec profil moléculaire. Le traitement implique une exérèse chirurgicale, une radiothérapie et, pour certaines tumeurs, une chimiothérapie. L'exérèse chirurgicale est rarement curative.
Les gliomes comprennent
Astrocytomes
Oligodendrogliomes
Glioblastome multiforme
Épendymomes
De nombreux gliomes infiltrent le tissu cérébral de manière diffuse et irrégulière.
Les gliomes à différentiation astrocytaire sont les gliomes les plus fréquents (voir aussi Astrocytomes chez l'enfant). Ils sont classés histologiquement et, dans certains cas, sur la base de la présence de marqueurs génétiques spécifiques, conformément à la classification de l'OMS (1).
Par ordre croissant de malignité, les astrocytomes sont classés en
Grade I: astrocytomes pilocytiques et astrocytomes sous-épendymaires à cellules géantes (le plus souvent dans la sclérose tubéreuse)
Grade II: astrocytomes de bas grade, y compris xanthoastrocytome pléomorphe
Grade III: astrocytomes anaplasiques
Grade IV: glioblastomes et gliomes médians diffus
Les astrocytomes pilocytaires, d'autres astrocytomes anaplasiques ou de bas grade ont tendance à se développer chez les patients jeunes. Les astrocytomes anaplasiques, en particulier, peuvent évoluer vers des glioblastomes (appelés glioblastomes secondaires). Les glioblastomes peuvent aussi se développer de novo (appelés glioblastomes primitifs), habituellement chez des personnes d'âge moyen ou âgées. Les glioblastomes contiennent des cellules ayant une hétérogénéïté chromosomique. Les glioblastomes primaires et secondaires ont des caractéristiques génétiques distinctes, qui peuvent changer à mesure que les tumeurs évoluent. Les glioblastomes secondaires portent généralement des mutations des gènes IDH1 ou IDH2.
Rarement, les astrocytomes contiennent des cellules d'astrocytome et d'oligodendrogliome. Ces tumeurs étaient appelées oligoastrocytomes; cependant, ce terme n'est plus utilisé pour désigner un seul type de tumeur, mais plutôt une néoplasie mixte.
Les oligodendrogliomes (grade II de l'OMS) sont généralement des gliomes à croissance la plus lente. Ils sont plus fréquents dans le cerveau antérieur, en particulier les lobes frontaux. Les oligodendrogliomes sont généralement caractérisés par une délétion du bras p du chromosome 1 et du bras q du chromosome 19 (codélétion 1p/19q). Ces délétions permettent de diagnostiquer les tumeurs oligodendrogliales, prédisent une survie plus longue et une meilleure réponse à la radiothérapie et à la chimiothérapie. Comme les astrocytomes, les oligodendrogliomes peuvent évoluer vers des formes plus agressives, telles que les oligodendrogliomes anaplasiques (grade III de l'OMS), qui sont pris en charge en conséquence.
Les astrocytomes et les oligodendrogliomes peuvent exprimer des mutations des gènes IDH1 ou IDH2 qui induisent une production anormale de 2-hydroxyglutarate; ce métabolite peut modifier la méthylation de l'ADN des cellules souches neurales et gliales normales et les amener à produire des cellules de gliome néoplasiques. Les patients qui ont une mutation IDH1/2 ont un meilleur pronostic que ceux qui ont des tumeurs de type IDH1/2 sauvages, en partie parce que les patients ont une meilleure réponse à la chimiothérapie alkylante telle que le témozolomide. Les oligodendrogliomes ont tendance à avoir une codélétion 1p/19q et une mutation IDH1/2. Les astrocytomes ont généralement des mutations IDH1/2 mais pas de codélétion 1p/19q; au lieu de cela, ils expriment plus généralement des mutations ou la perte du gène ATRX et des mutations de pTP53 (2).
Les gliomes médians diffus sont des tumeurs astrocytaires de haut grade (grades III à IV de l'OMS) qui affectent principalement les enfants. Ces tumeurs comprennent des gliomes pontins intrinsèques diffus, des tumeurs agressives et typiquement létales qui infiltrent le tronc cérébral avec extension rostrale dans l'hypothalamus et le thalamus et infiltrent le bulbe et la moelle épinière par le bas. Les gliomes médians diffus expriment généralement la mutation H3K27M.
Les enfants atteints de neurofibromatose de type 1 présentent un risque accru de développer des gliomes diffus de la ligne médiane.
Les épendymomes surviennent principalement chez l'enfant et le jeune adulte; ils sont rares après l'adolescence (voir aussi Ependymomes chez l'enfant). Ils sont classés comme suit
Grade I: sous-épendymome
Grade II: épendymome
Grade III: épendymome anaplasique
Grade IV: épendymoblastomes (ils sont rares et surviennent principalement chez les nourrissons)
Tous les épendymomes proviennent généralement de la paroi ventriculaire et peuvent donc se former dans le cerveau, le tronc cérébral ou la moelle épinière. Ils sont donc classés en fonction de leur emplacement: comme supratentoriels, de la fosse postérieure ou médullaires. Chacune de ces catégories comprend 3 sous-ensembles définis moléculairement et histologiquement, aboutissant à 9 types d'épendymomes phénotypiquement et moléculairement distincts, dont le traitement et le pronostic diffèrent significativement l'un de l'autre. Les épendymomes du 4ème ventricule en particulier peuvent se manifester par une hydrocéphalie obstructive et peut donc se manifester plus tôt que d'autres épendymomes (3).
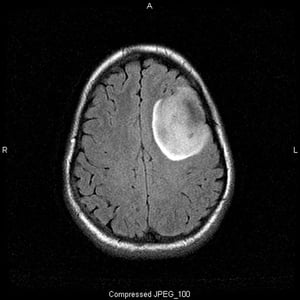
Image courtesy of William R. Shapiro, MD.
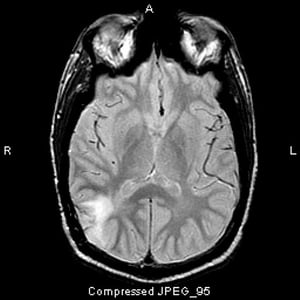
Image courtesy of William R. Shapiro, MD.
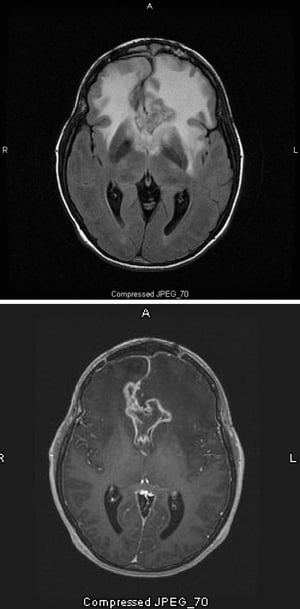
Images courtoisie de William R. Shapiro, MD.

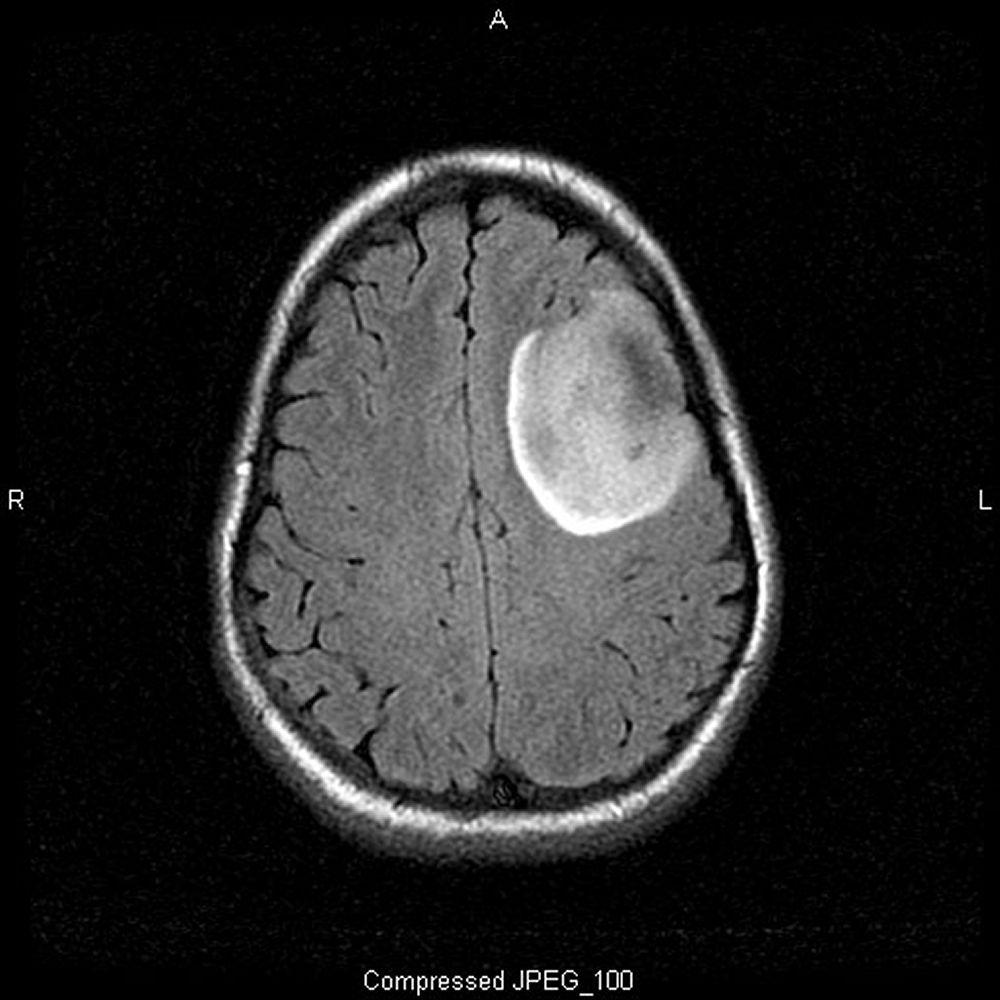
Image courtesy of William R. Shapiro, MD.
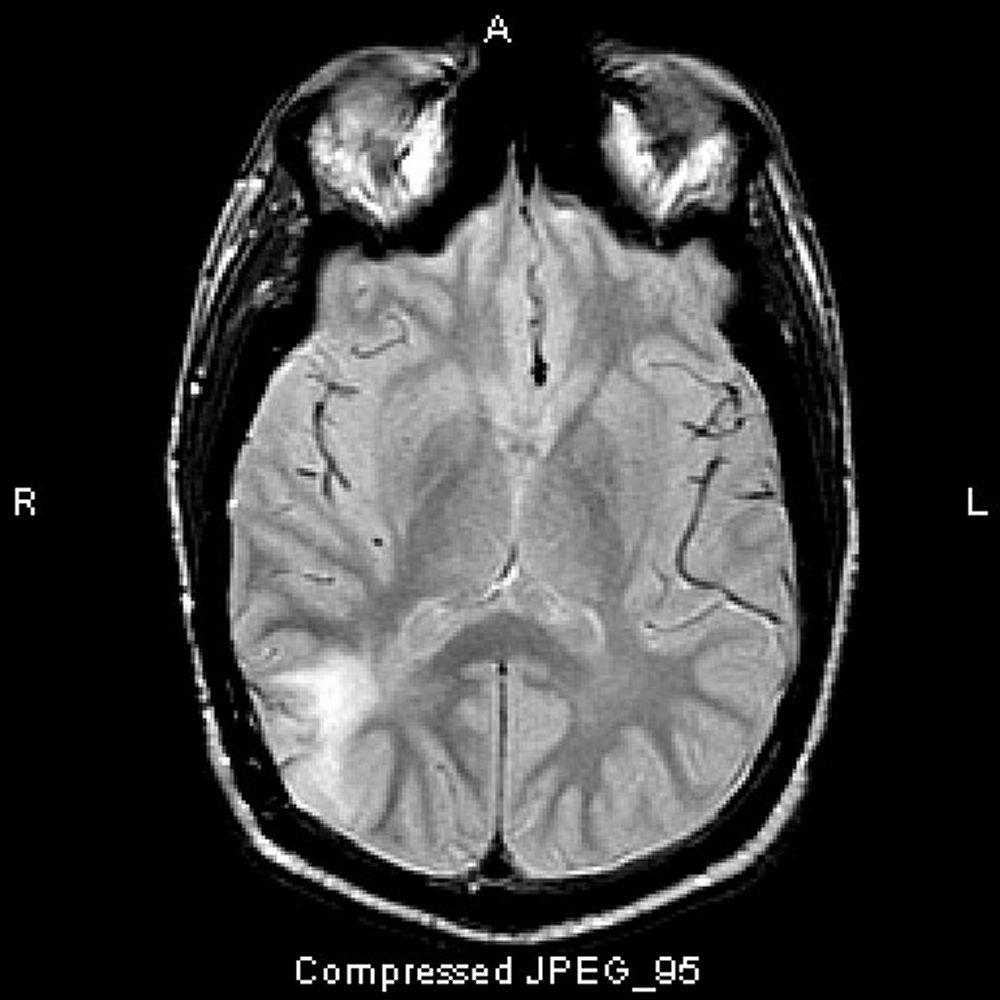
Image courtesy of William R. Shapiro, MD.

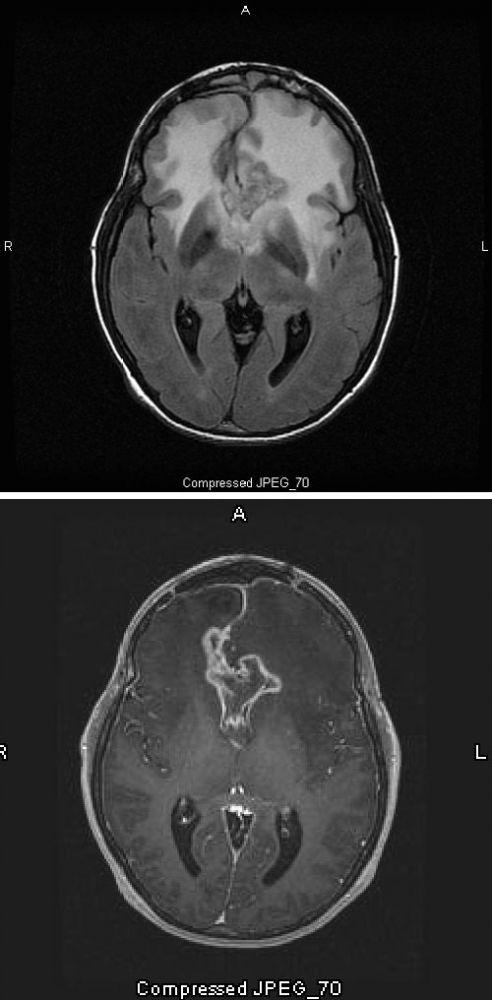
Images courtoisie de William R. Shapiro, MD.
Références générales
1. Louis DN, Perry A, Reifenberger G, et al: The 2021 World Health Organization classification of tumors of the central nervous system: A summary. Neuro Oncol 23 (8):1231–1251, 2021. doi: 10.1093/neuonc/noab106
2. Reifenberger G, Wirsing H, Knobbe-Thomsen CB, Weller M: Advances in the molecular genetics of gliomas - implications for classification and therapy. Nat Rev Clin Oncol 14 (7):434-452 2017. doi: 10.1038/nrclinonc.2016.204
3. Pajtler K, Mack S, Ramaswamy V, et al: The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Neuropathologica 133:5–12, 2017. doi: 10.1007/s00401-016-1643-0
Diagnostic des gliomes
IRM pondérées en T1 et T2
Biopsie
Le diagnostic de gliomes repose principalement sur l'IRM, qui comprend l'imagerie standard pondérée en T1 et en T2, de préférence avec rehaussement au gadolinium, suivie d'une biopsie avec histopathologie et profil moléculaire. L'histopathologie comprend l'analyse anatomopathologique classique de la morphologie cellulaire, les colorations immunohistochimiques (p. ex., des mutations d'IDH1/2), et l'hybridation in situ (p. ex., pour les mutations d'EGFR).
Des panels d'oncogènes sélectionnés, un séquençage ciblé ou de l'exome entier, un séquençage de l'ARN et/ou une analyse de méthylation de la méthylguanine-ADN méthyl-transférase (MGMT) peuvent être effectués. La méthylation du promoteur du gène MGMT est un facteur pronostique en cas de glioblastome multiforme; il prédit une bonne réponse au témozolamide post-opératoire et une survie accrue (1).
Référence pour le diagnostic
1. Stupp R, Taillibert S, Kanner A, et al: Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 318 (23):2306–2316, 2017. doi: 10.1001/jama.2017.18718
Traitement des gliomes
Exérèse chirurgicale
Radiothérapie
Chimiothérapie pour certains types
Astrocytomes anaplasiques et glioblastomes
Le traitement implique la chirurgie, la radiothérapie, la chimiothérapie pour réduire le volume tumoral. Exciser en sécurité autant de tumeur que possible (sans léser des zones fondamentales du cerveau [les zones qui gèrent des fonctions telles que la parole et la fonction motrice]) prolonge la survie et améliore la fonction neurologique.
Après l'intervention, les patients reçoivent une radiothérapie à dose pleine (60 Gy [gray] sur 6 semaines); idéalement, une radiothérapie conformationnelle, qui cible la tumeur et épargne le tissu cérébral normal.
Dans le cas des glioblastomes, la chimiothérapie par le témozolomide est désormais administrée systématiquement avec la radiothérapie. La dose est
75/mg/m2/jour pendant 42 jours (dont les week-ends où l'irradiation est omise)
Puis 150 mg/m2 par voie orale 1 fois/jour pendant 5 jours/mois au cours du mois suivant
Suivi de 200 mg/m2 par voie orale 1 fois/jour pendant 5 jours/mois les mois suivants pour un total de 6 à 12 mois
Pendant le traitement par témozolomide une association triméthoprime/sulfaméthoxazole 800 mg/160 mg peut être administrée 3 fois/semaine pour prévenir une pneumonie à Pneumocystis jirovecii.
Les patients recevant une chimiothérapie ont besoin d'une NFS à intervalles réguliers.
Une implantation de disques résorbables de chimiothérapie au cours de la résection chirurgicale peut être indiquée chez certains patients.
L'utilisation de champs de traitement tumoraux avec du témozolomide en adjuvant peut être adapté au cas de certains patients. Les champs de traitement des tumeurs perturbent avec les mitoses du glioblastome et l'assemblage des organites en transmettant des champs électriques alternatifs au cuir chevelu. Dans un essai clinique randomisé, les champs de traitement des tumeurs ont semblé améliorer la survie des patients atteints de glioblastome (1).
Les protocoles expérimentaux (p. ex., radiochirurgie stéréotaxique, nouveaux médicaments, thérapie génique ou immunothérapie) doivent être également envisagés. Des essais cliniques de nombreux inhibiteurs des points de contrôle immunitaires (checkpoint inhibitors), en particulier de la modulation immunitaire dépendante de CTLA4 et PD1, sont en cours.
Après un traitement multimodal conventionnel, les taux de survie en cas de glioblastomes sont d'environ 50% à 1 an, 25% à 2 ans et de 10 à 15% à 5 ans. Le pronostic est meilleur dans les cas suivants:
Si les patients ont < 45 ans.
L'histologie est celle d'un astrocytome anaplasique ou d'une tumeur de grade inférieur (plutôt que d'un glioblastome).
Si l'exérèse initiale améliore la fonction neurologique et laisse peu ou pas de tumeur résiduelle.
Les tumeurs ont une mutation IDH1.
La méthylation du promoteur MGMT (méthylguanine méthyltransférase) est présente.
Avec un traitement classique, le temps de survie médian est d'environ 30 mois en cas d'astrocytome anaplasique et d'environ 15 mois en cas de glioblastome. Un large éventail d'essais cliniques, en particulier ceux d'un certain nombre d'immunothérapies expérimentales et d'inhibiteurs de point de contrôle immunitaire, sont en cours. Les essais en cours et récemment terminés sont référencés dans clinical trials.gov.
Astrocytomes et oligodendrogliomes de bas grade
La résection chirurgicale maximale sans danger est indiquée pour les astrocytomes et les oligodendrogliomes de bas grade. Après résection complète chez les patients de < 40 ans, une observation peut être envisagée. Pour les autres patients, la radiothérapie et la chimiothérapie adjuvante prolongent la survie (2).
La médiane de survie varie de 1 à 2 ans chez les patients à haut risque (sans mutation IDH1, avec résection incomplète) à > 10 ans chez ceux qui ont des facteurs pronostiques favorables. Chez les patients à haut risque, le cancer est susceptible de progresser.
Gliomes médians diffus
Chez les patients présentant des gliomes de la ligne médiane diffus, la radiothérapie peut ralentir la progression de la maladie, bien que son utilisation soit principalement palliative, car la survie est généralement inférieure à un an. Ces tumeurs expriment généralement la mutation H3K27M d'une protéine histone; des stratégies épigénétiques qui modifient l'ADN et les protéines associées à l'ADN de la cellule pour modifier l'expression des gènes sont à l'étude (3). Les patients porteurs de la mutation H3K27M ont un mauvais pronostic quel que soit le grade histologique de la tumeur.
Épendymomes
La biopsie avec analyse génétique, le prélèvement de liquide céphalorachidien et l'imagerie craniospinale doivent être utilisés pour classer les épendymomes et évaluer leur propagation dans le système nerveux central.
Le traitement des épendymomes comprend une résection chirurgicale maximale sans danger en cas de maladie unifocale ou de tumeurs symptomatiques. Pour les tumeurs de grade supérieur, la radiothérapie peut être dirigée localement ou inclure tout l'axe craniospinal en fonction de l'étendue de la propagation de la tumeur. Le rôle de la chimiothérapie n'est pas bien défini.
Le pronostic varie selon la localisation, la génétique et le stade de la tumeur (4). En général, grâce au traitement, la survie globale à 5 ans est d'environ 50%; cependant, chez les patients qui n'ont pas de tumeur résiduelle, la survie à 5 ans est > 70%.
Références pour le traitement
1. Stupp R, Taillibert S, Kanner A, et al: Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 318 (23):2306–2316, 2017. doi: 10.1001/jama.2017.18718
2. Buckner JC, Shaw EG, Pugh SL, et al: Radiation plus procarbazine, CCNU, and vincristine in low-grade glioma. N Engl J Med 374 (14):1344-1355, 2016. doi: 10.1056/NEJMoa1500925
3. Miklja Z, Pasternak A, Stallard S, et al: Molecular profiling and targeted therapy in pediatric gliomas: Review and consensus recommendations. Neuro Oncol 21:968–980, 2019.
4. Pajtler K, Mack S, Ramaswamy V, et al: The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Neuropathologica 133:5–12, 2017. doi: 10.1007/s00401-016-1643-0
Points clés
Les gliomes sont des tumeurs primitives qui ont pour origine le parenchyme cérébral; ils comprennent les astrocytomes, les oligodendrogliomes et les épendymomes.
Les gliomes varient selon le site, le degré de malignité, le traitement et le pronostic.
Pour la plupart des gliomes, exciser chirurgicalement autant de tumeurs que possible (résection totale brute) sans léser indûment les zones de cerveau vitales, puis poursuivre par radiothérapie et/ou chimiothérapie.







