

Multiple endocrine neoplasia, type 2B (MEN 2B) is an autosomal dominant syndrome characterized by medullary thyroid carcinoma, pheochromocytoma, multiple mucosal neuromas and intestinal ganglioneuromas, and often a marfanoid habitus and other skeletal abnormalities. Symptoms depend on the glandular abnormalities present. Diagnosis is confirmed by genetic testing. Hormonal and imaging tests help locate the tumors, which are removed surgically when possible.
(See also Overview of Multiple Endocrine Neoplasias.)
Ninety-five percent of MEN 2B cases result from a single amino acid substitution at amino acid 918 in the RET protein. As in MEN 2A and familial medullary thyroid carcinoma, this mutation results in activation of RET proto-oncogene–mediated cellular processes. More than 50% are de novo mutations and thus may be sporadic rather than familial.
Symptoms and Signs of MEN 2B
Symptoms and signs reflect the glandular abnormalities present (see table ). Approximately 50% of patients have the complete syndrome with mucosal neuromas, pheochromocytomas, and medullary thyroid carcinoma. Fewer than 10% have neuromas and pheochromocytomas alone, whereas the remaining patients have neuromas and medullary thyroid carcinoma without pheochromocytoma.

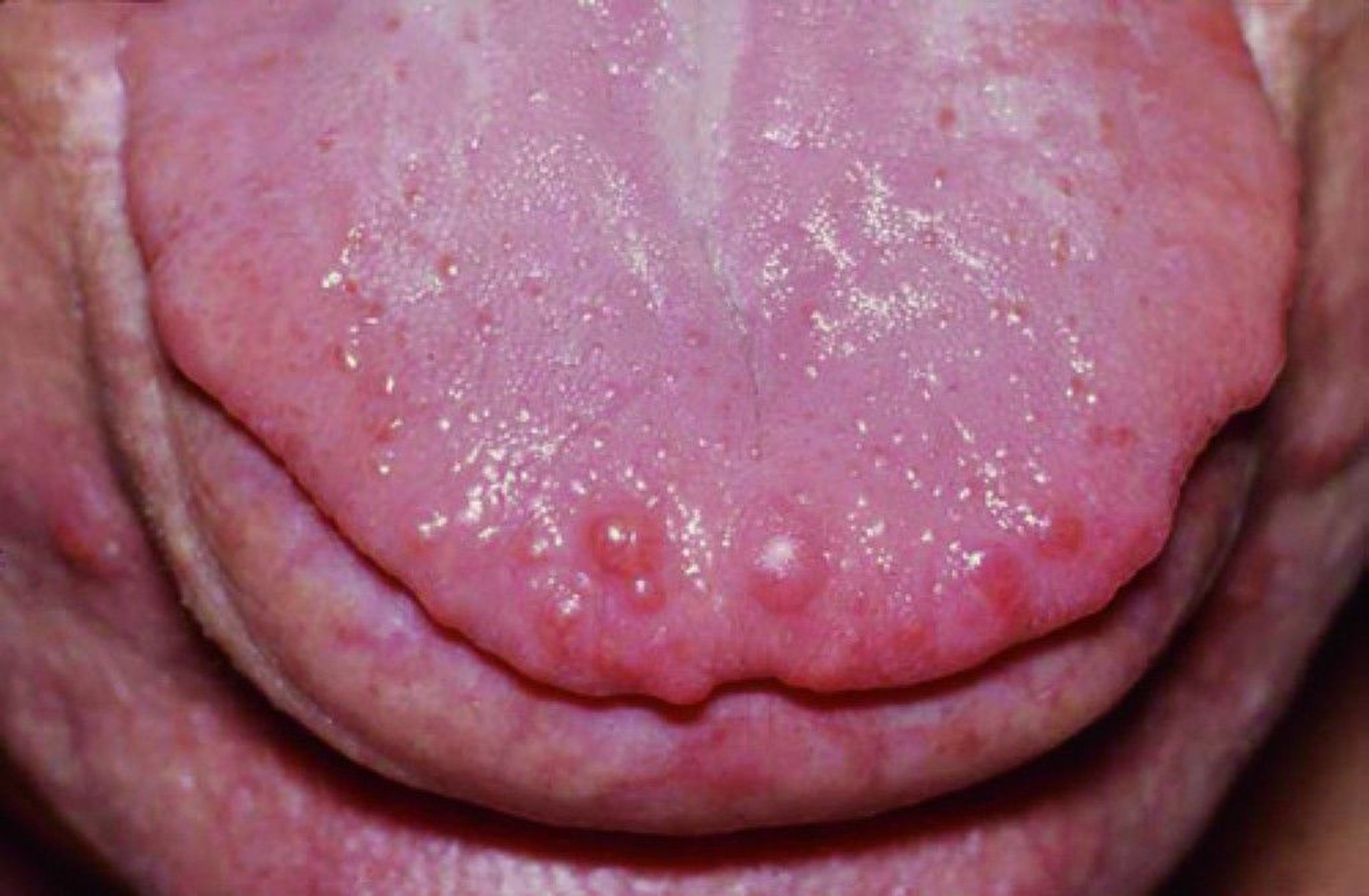
Mucosal neuromas may appear as small glistening bumps on the tongue.
© Springer Science+Business Media
Often, mucosal neuromas are the earliest sign, and they occur in most or all patients. Neuromas appear as small glistening bumps on the lips, tongue, and buccal mucosae.
The eyelids, conjunctivae, and corneas also commonly develop neuromas; infants are often unable to make tears. Thickened eyelids and everted, diffusely hypertrophied lips are characteristic.
Gastrointestinal abnormalities related to altered motility (constipation, diarrhea, and, occasionally, megacolon) are common and thought to result from diffuse intestinal ganglioneuromatosis.
Patients almost always have a marfanoid habitus. Skeletal abnormalities are common, including deformities of the spine (lordosis, kyphosis, scoliosis), slipped capital femoral epiphyses, dolichocephaly (hull-shaped skull, also called scaphocephaly), pes cavus, and talipes equinovarus.
Medullary thyroid carcinoma and pheochromocytoma resemble the corresponding disorders in MEN 2A syndrome; both tend to be bilateral and multicentric. Medullary thyroid carcinoma, however, tends to be particularly aggressive in MEN 2B and is almost always present by 1 year of age.
Although the neuromas, facial characteristics, and gastrointestinal disorders are present at an early age, the syndrome may not be recognized until medullary thyroid carcinoma or pheochromocytoma manifests in later life.
Diagnosis of MEN 2B
Serum calcitonin levels
Plasma free metanephrines and urinary catecholamine levels
Neck CT or MRI
Pheochromocytoma localization with MRI or CT
Genetic testing
MEN 2B is suspected in patients with a family history of MEN 2B, pheochromocytoma, multiple mucosal neuromas, or medullary thyroid carcinoma. Genetic testing is highly accurate and is done to confirm the disorder. Genetic testing also is done to screen first-degree relatives and any symptomatic family members of patients with MEN 2B, as in MEN 2A.
Pheochromocytoma may be suspected clinically and is confirmed by measuring plasma free metanephrines and urinary catecholamines.
Laboratory testing for medullary thyroid carcinoma with serum calcitonin measurements should be done.
MRI or CT is used to search for pheochromocytomas and medullary thyroid carcinoma.
Treatment of MEN 2B
Surgical excision of identified tumors
Prophylactic thyroidectomy
Affected patients should have total thyroidectomy as soon as the diagnosis is established. Pheochromocytoma, if present, should be removed before thyroidectomy is done.
Gene carriers should undergo prophylactic thyroidectomy before age 1 year.
Key Points
Multiple endocrine neoplasia, type 2B (MEN 2B) has a mutation of the same gene as in MEN 2A and manifests similarly except for the absence of hyperparathyroidism, the presence of more aggressive medullary thyroid carcinoma and the presence of multiple mucosal neuromas and a marfanoid habitus.
Patients should have genetic testing for RET proto-oncogene mutations, serum calcitonin measurement, blood or urine tests for pheochromocytoma, and imaging studies of the neck and adrenal glands.
Pheochromocytoma is excised and prophylactic thyroidectomy is done.





