

Alcohol consumption is high in most Western countries. According to the United States-based 2023 National Survey on Drug Use and Health, which uses the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) definition of alcohol use disorder, 10.9% of adults in the United States age 18 and older and 2.9% of children age 12 to 17 met criteria for alcohol use disorder in 2022 (1; National Institute on Alcohol Abuse and Alcoholism [NIAAA]: Alcohol Use Disorder (AUD) in the United States). The NIAAA reported a 33% increase in deaths involving alcohol use, from approximately 79,000 in 2019 to 105,000 in 2022. This represents an average annual increase of approximately 10%, up from the 2.2% annual increase over the previous two decades, suggesting a a potential relationship to the COVID-19 pandemic (2, 3).
Disorders of the liver that occur in people with alcohol use disorder, often in sequence, but sometimes coexisting, include (4):
Alcohol-associated hepatic steatosis (fatty liver; in > 90%)
Alcoholic hepatitis (in 10 to 35%)
Alcohol-associated cirrhosis (in 10 to 20%)
Hepatocellular carcinoma may also develop in patients with cirrhosis, especially if iron accumulation coexists.
References
1. SAMHSA, Center for Behavioral Health Statistics and Quality. 2022 National Survey on Drug Use and Health. Table 5.9A—Alcohol use disorder in past year: among people aged 12 or older; by age group and demographic characteristics, numbers in thousands, 2022 and 2023. Accessed March 19, 2025.
2. White AM, Castle IP, Powell PA, et al. Alcohol-Related Deaths During the COVID-19 Pandemic. JAMA. 327(17):1704-1706, 2022. doi:10.1001/jama.2022.4308
3. Centers for Disease Control and Prevention, National Center for Health Statistics. National Vital Statistics System, Provisional Mortality on CDC WONDER Online Database. Data are from the final Multiple Cause of Death Files, 2018-2022, and from provisional data for years 2023-2024, as compiled from data provided by the 57 vital statistics jurisdictions through the Vital Statistics Cooperative Program. Accessed March 19, 2025.
4. Crabb DW, Im GY, Szabo G, et al. Diagnosis and Treatment of Alcohol-Associated Liver Diseases: 2019 Practice Guidance From the American Association for the Study of Liver Diseases. Hepatology. 2020;71(1):306-333. doi:10.1002/hep.30866
Risk Factors for Alcohol-Related Liver Disease
The main risk factors for alcohol-related liver disease are:
Quantity and duration of alcohol use
Sex
Genetic and metabolic traits
Obesity
History of bariatric surgery
Quantity of alcohol
A standard alcoholic drink contains 14 grams of alcohol, the amount found in a 12-ounce (355-mL) bottle of 5% beer, a 5-ounce (148-mL) glass of wine, or 1.5 ounces (44-mL) of a beverage of 80-proof distilled spirits containing 40% alcohol by volume (1).
There appears to be a threshold effect above which the amount and duration of alcohol use increases the risk of the development of liver disease. That threshold is unknown and varies by individual risk factors (2).
The NIAAA defines drinking in moderation as one standard drink per day for women and two standard drinks per day for men (1, 3 ). However, the World Health Organization states that there is no safe level of alcohol consumption that does not affect health (4).
The NIAAA defines "at-risk" (heavy) drinking in males as more than 14 standard drinks per week, or more than 4 drinks per day, and in females as more than 7 standard drinks per week, or more than 3 drinks per day (1).
Binge drinking may also increase alcohol-related liver disease. The NIAAA defines binge drinking as a pattern of drinking that brings blood alcohol concentration levels to 0.08 g/dL, which typically occurs after 4 drinks for women and 5 drinks for men, in about 2 hours (1).
Patients with alcohol-related liver disease or other liver diseases, in particular metabolic dysfunction-associated steatotic liver disease (MASLD) (previously called nonalcoholic fatty liver disease [NAFLD]), metabolic dysfunction-associated steatohepatitis (MASH) (previously called nonalcoholic steatohepatitis [NASH]), viral hepatitis, and hemochromatosis, or any drinking that leads to negative consequences, should be counseled that there is no safe level of drinking and that they should abstain.
The American Association for the Study of Liver Diseases (AASLD) and American College of Gastroenterology recommend that patients receiving care in primary care and gastroenterology/hepatology outpatient clinics, emergency departments, and in hospitals (as inpatients) be screened routinely for alcohol use with validated questionnaires. Brief intervention, pharmacotherapy, and referral to treatment should be offered to patients engaged in hazardous drinking (ie, heavy or binge drinking) (5-7). The AASLD recommends using the Alcohol Use Disorders Inventory Test (AUDIT) if excessive alcohol use is suspected (5).
Alcohol biomarkers, such as urine or hair ethyl glucuronide, urine ethyl sulfate, and phosphatidylethanol (PEth), can be used to support patient history and aid in recovery.
Sex
Women are more susceptible to alcohol-related liver disease, even after adjustment for body size. Women may require only 20 to 30 g of alcohol/day to be at risk—half or less than the amount for men (7, 8). Risk in women may be increased because they have less alcohol dehydrogenase in their gastric mucosa; thus, more intact alcohol reaches the liver.
Genetic factors
Alcohol-related liver disease often runs in families, suggesting genetic factors. Some specific genes (eg, deficiency of cytoplasmic enzymes that eliminate alcohol) have been identified and are under investigation (7).
Nutrition or obesity
A diet high in unsaturated fat increases susceptibility. Obesity alone or with a history of bariatric surgery is a risk factor (9).
Other factors
Other risk factors include iron accumulation in the liver (not necessarily related to iron intake) and concomitant viral hepatitis.
Risk factors references
1. Alcohol Research: Current Reviews Editorial Staff. Drinking Patterns and Their Definitions. Alcohol Res. 2018;39(1):17-18.
2. Rehm J, Taylor B, Mohapatra S, et al. Alcohol as a risk factor for liver cirrhosis: a systematic review and meta-analysis. Drug Alcohol Rev. 29(4):437-445, 2010. doi:10.1111/j.1465-3362.2009.00153.x
3. U.S. Department of Agriculture and U.S. Department of Health and Human Services. Dietary Guidelines for Americans, 2020-2025. 9th Edition. December 2020. Available at DietaryGuidelines.gov.
4. Anderson BO, Berdzuli N, Ilbawi A, et al. Health and cancer risks associated with low levels of alcohol consumption. Lancet Public Health. 8(1):e6-e7, 2023. doi: 10.1016/S2468-2667(22)00317-6
5. Crabb DW, Im GY, Szabo G, et al. Diagnosis and Treatment of Alcohol-Associated Liver Diseases: 2019 Practice Guidance From the American Association for the Study of Liver Diseases. Hepatology. 2020;71(1):306-333. doi:10.1002/hep.30866
6. Saunders JB, Aasland OG, Babor TF, et al. Development of the Alcohol Use Disorders Identification Test (AUDIT): WHO Collaborative Project on Early Detection of Persons with Harmful Alcohol Consumption--II. Addiction. 88(6):791-804, 1993. doi:10.1111/j.1360-0443.1993.tb02093.x
7. Jophlin LL, Singal AK, Bataller R, et al. ACG Clinical Guideline: Alcohol-Associated Liver Disease. Am J Gastroenterol. 2024;119(1):30-54. doi:10.14309/ajg.0000000000002572
8. O'Shea RS, Dasarathy S, McCullough AJ; Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease. Hepatology. 2010;51(1):307-328. doi:10.1002/hep.23258
9. Mellinger JL, Shedden K, Winder GS, et al. Bariatric surgery and the risk of alcohol-related cirrhosis and alcohol misuse. Liver Int. 2021;41(5):1012-1019. doi:10.1111/liv.14805
Pathophysiology of Alcohol-Related Liver Disease
Alcohol absorption and metabolism
Alcohol (ethanol) is readily absorbed from the stomach, but most is absorbed from the small intestine. Alcohol cannot be stored. A small amount is degraded in transit through the gastric mucosa, but most is catabolized in the liver, primarily by alcohol dehydrogenase (ADH) but also by cytochrome P-450 2E1 (CYP2E1) and the microsomal enzyme oxidation system (MEOS).Alcohol (ethanol) is readily absorbed from the stomach, but most is absorbed from the small intestine. Alcohol cannot be stored. A small amount is degraded in transit through the gastric mucosa, but most is catabolized in the liver, primarily by alcohol dehydrogenase (ADH) but also by cytochrome P-450 2E1 (CYP2E1) and the microsomal enzyme oxidation system (MEOS).

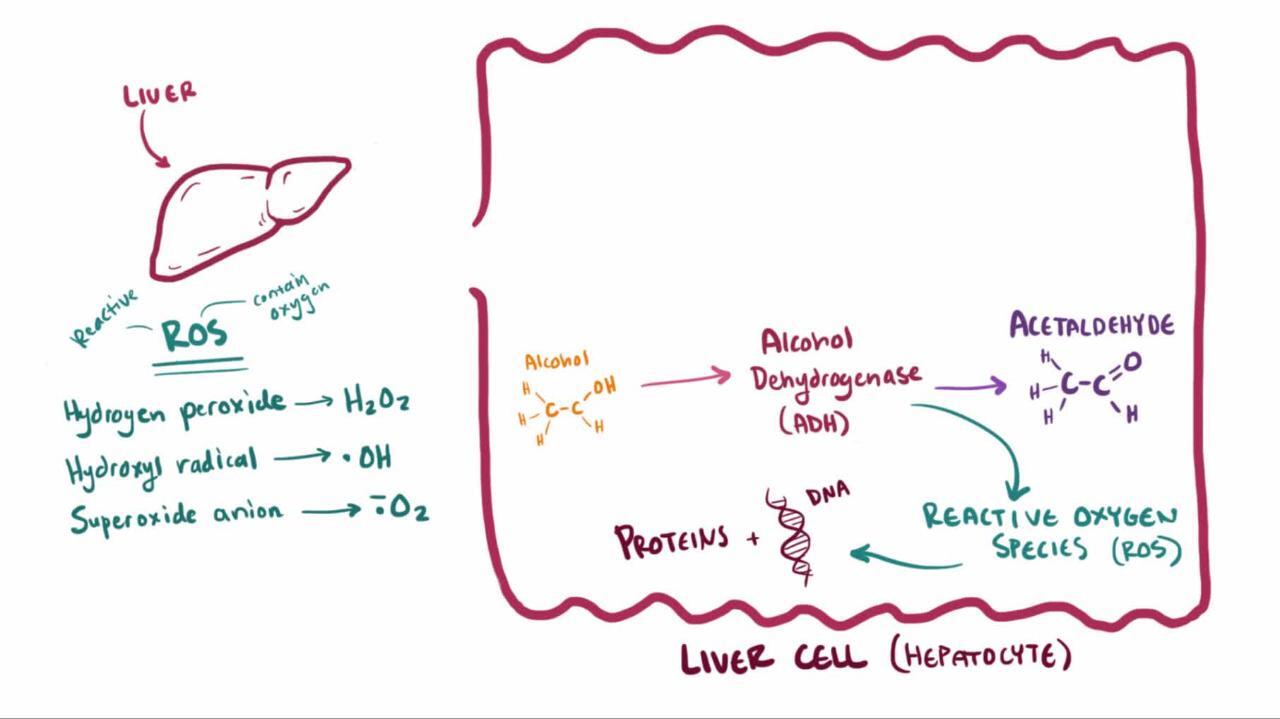
Metabolism via the ADH pathway involves the following:
ADH, a cytoplasmic enzyme, oxidizes alcohol into acetaldehyde. Genetic polymorphisms in ADH account for some individual differences in blood alcohol levels after the same alcohol intake but not in susceptibility to alcohol-related liver disease.
Acetaldehyde dehydrogenase (ALDH), a mitochondrial enzyme, then oxidizes acetaldehyde into acetate. Chronic alcohol consumption enhances acetate formation. People of East Asian descent often have lower levels of ALDH due to an allelic variation and are more susceptible to toxic acetaldehyde effects (eg, flushing); the effects are similar to those of disulfiram, which inhibits ALDH (Acetaldehyde dehydrogenase (ALDH), a mitochondrial enzyme, then oxidizes acetaldehyde into acetate. Chronic alcohol consumption enhances acetate formation. People of East Asian descent often have lower levels of ALDH due to an allelic variation and are more susceptible to toxic acetaldehyde effects (eg, flushing); the effects are similar to those of disulfiram, which inhibits ALDH (1).
These oxidative reactions generate hydrogen, which converts nicotinamide-adenine dinucleotide (NAD) to its reduced form (NADH), increasing the redox potential (NADH/NAD) in the liver.
The increased redox potential inhibits fatty acid oxidation and gluconeogenesis, promoting fat accumulation in the liver.
Chronic excessive alcohol consumption induces the MEOS (mainly in endoplasmic reticulum), increasing its activity. The main enzyme involved is CYP2E1. When induced, the MEOS pathway can account for 20% of alcohol metabolism (2). This pathway generates harmful reactive oxygen species, increasing oxidative stress and formation of oxygen-free radicals.
Hepatic fat accumulation
Fat (triglycerides) accumulates throughout the hepatocytes for the following reasons:
Export of fat from the liver is decreased because hepatic fatty acid oxidation and lipoprotein production decrease.
Input of fat is increased because the decrease in hepatic fat export increases peripheral lipolysis and triglyceride synthesis, resulting in hyperlipidemia.
Hepatic fat accumulation may predispose to subsequent oxidative damage.
Endotoxins in the gut
Alcohol changes gut permeability, increasing absorption of endotoxins released by bacteria in the gut. In response to the endotoxins (which the impaired liver can no longer detoxify), liver macrophages (Kupffer cells) release free radicals, increasing oxidative damage.
Oxidative damage
Oxidative stress is increased by:
Liver hypermetabolism, caused by alcohol consumption
Free radical–induced lipid peroxidative damage
Reduction in protective antioxidants (eg, glutathione, vitamins A and E), caused by alcohol-related undernutrition
Binding of alcohol oxidation products, such as acetaldehyde, to liver cell proteins, forming neoantigens and resulting in inflammation
Accumulation of neutrophils and other white blood cells (WBCs), which are attracted by lipid peroxidative damage and neoantigens
Inflammatory cytokines secreted by WBCs
Accumulation of hepatic iron, if present, aggravates oxidative damage. Iron can accumulate in alcohol-related liver disease through ingestion of iron-containing fortified wines; most often, the iron accumulation is modest. This condition can be differentiated from hereditary hemochromatosis.
Resultant inflammation, cell death, and fibrosis
A vicious circle of worsening inflammation occurs: Cell necrosis and apoptosis result in hepatocyte loss, and subsequent attempts at regeneration result in fibrosis. Stellate (Ito) cells, which line blood channels (sinusoids) in the liver, proliferate and transform into myofibroblasts, producing an excess of type I collagen and extracellular matrix. As a result, the sinusoids narrow, limiting blood flow. Fibrosis narrows the terminal hepatic venules, compromising hepatic perfusion and thus contributing to portal hypertension. Extensive fibrosis is associated with an attempt at regeneration, resulting in liver nodules. This process culminates in cirrhosis.
Pathophysiology references
1. Yin SJ, Peng GS. Overview of ALDH polymorphism: relation to cardiovascular effects of alcohol. In Preedy VR, Watson RR, eds. Comprehensive Handbook of Alcohol Related Pathology, Vol. 1. (VR Preedy, RR Watson eds), pp 409–424. Elsevier; 2005:409-424.
2. Contreras-Zentella ML, Villalobos-García D, Hernández-Muñoz R. Ethanol Metabolism in the Liver, the Induction of Oxidant Stress, and the Antioxidant Defense System. . Ethanol Metabolism in the Liver, the Induction of Oxidant Stress, and the Antioxidant Defense System.Antioxidants (Basel). 2022;11(7):1258. Published 2022 Jun 26. doi:10.3390/antiox11071258
Pathology of Alcohol-Related Liver Disease
Hepatic steatosis, alcoholic hepatitis, and cirrhosis are often considered separate, progressive manifestations of alcohol-related liver disease. However, their features often overlap.
Alcohol-related hepatic steatosis (fatty liver) is the initial and most common consequence of excessive alcohol consumption. Hepatic steatosis is potentially reversible. Macrovesicular fat accumulates as large droplets of triglyceride and displaces the hepatocyte nucleus, most markedly in perivenular hepatocytes. The liver enlarges.
Alcoholic hepatitis (steatohepatitis) is a combination of hepatic steatosis, diffuse liver inflammation, and liver necrosis (often focal)—all in various degrees of severity. The damaged hepatocytes are swollen with a granular cytoplasm (balloon degeneration) or contain fibrillar protein in the cytoplasm (Mallory or alcoholic hyaline bodies). Severely damaged hepatocytes become necrotic. Sinusoids and terminal hepatic venules are narrowed. Cirrhosis may also be present.
Alcohol-related cirrhosis is advanced liver disease characterized by extensive fibrosis that disrupts the normal liver architecture. The amount of fat present varies. Alcoholic hepatitis may coexist. The feeble compensatory attempt at hepatic regeneration produces relatively small nodules (micronodular cirrhosis). As a result, the liver usually shrinks. In time, even with abstinence, fibrosis forms broad bands, separating liver tissue into large nodules (macronodular cirrhosis—see Cirrhosis: Pathophysiology).
Symptoms and Signs of Alcohol-Related Liver Disease
Symptoms usually become apparent in patients during their 30s or 40s; severe problems appear approximately a decade later.
Alcohol-related hepatic steatosis is often asymptomatic. In one third of patients, the liver is enlarged and smooth, but it is not usually tender.


Light micrograph of a section of liver tissue affected by steatosis (fatty liver). This condition leads to the accumulation of triglycerides (which appear as white spaces in micrograph) in liver cells due to abnormal lipid metabolism.
Haematoxylin-eosin saffron (HES) stain. Magnification: 556x when printed at 10 centimetres.
PROF. J.L. KEMENY / SCIENCE PHOTO LIBRARY
Alcoholic hepatitis ranges from mild and reversible to life threatening. Most patients with moderate disease are undernourished and present with fatigue, fever, jaundice, right upper quadrant pain, tender hepatomegaly, and sometimes a hepatic bruit. Approximately 40% deteriorate soon after hospitalization, with consequences ranging from mild (eg, increasing jaundice) to severe (eg, ascites, portosystemic encephalopathy, variceal bleeding, liver failure with hypoglycemia, coagulopathy). Other manifestations of cirrhosis may be present.
Cirrhosis, if compensated, may be asymptomatic. The liver is usually small; when the liver is enlarged, hepatic steatosis or hepatoma should be considered. Symptoms range from those of alcoholic hepatitis to the complications of end-stage liver disease, such as portal hypertension (often with esophageal varices and upper gastrointestinal bleeding, splenomegaly, ascites, and portosystemic encephalopathy). Portal hypertension may lead to intrapulmonary arteriovenous shunting with hypoxemia (hepatopulmonary syndrome), which may cause cyanosis and nail clubbing. Acute renal failure secondary to progressively decreasing renal blood flow (hepatorenal syndrome) may develop. Hepatocellular carcinoma develops in 10 to 15% of patients with alcohol-related cirrhosis.


Light micrograph of a human liver with micronodular cirrhosis, a frequent consequence of alcoholic hepatitis. The regenerating nodules are separated by connective tissue septa showing chronic inflammatory infiltrates. Note the absence of a central vein in the nodules and of portal triads in the connective tissue septa.
JOSE CALVO / SCIENCE PHOTO LIBRARY
Chronic excessive alcohol consumption, rather than liver disease, causes Dupuytren contracture of the palmar fascia, vascular spiders, myopathy, and peripheral neuropathy. In men, chronic excessive alcohol consumption causes signs of hypogonadism and feminization (eg, smooth skin, lack of male-pattern baldness, gynecomastia, testicular atrophy, decreased body hair). Undernutrition may lead to multiple vitamin deficiencies (eg, of folate and thiamin), enlarged parotid glands, and white nails. In those with chronic excessive alcohol consumption, Wernicke encephalopathy and Korsakoff psychosis result mainly from thiamine deficiency. Pancreatitis is common. Hepatitis C occurs in > 16 to 24% of those with alcohol use disorder (1, 2); this combination markedly worsens the progression of liver disease.
Rarely, patients with hepatic steatosis or cirrhosis present with Zieve syndrome (hyperlipidemia, hemolytic anemia, and jaundice).


DR P. MARAZZI/SCIENCE PHOTO LIBRARY
Symptoms and signs references
1. Novo-Veleiro I, Calle Cde L, Domínguez-Quibén S, et al. Prevalence of hepatitis C virus infection in alcoholic patients: cohort study and systematic review. Alcohol Alcohol. 2013;48(5):564-569. doi:10.1093/alcalc/agt044
2. Laskus T, Radkowski M, Lupa E, et al. Prevalence of markers of hepatitis viruses in out-patient alcoholics. J Hepatol. 1992;15(1-2):174-178. doi:10.1016/0168-8278(92)90032-k
Diagnosis of Alcohol-Related Liver Disease
Confirmed history of alcohol use
Alcohol biomarkers
Liver tests and complete blood count (CBC)
Sometimes liver biopsy
Alcohol is suspected as the cause of liver disease in any patient who chronically consumes excess alcohol, particularly > 80 g per day. Patients can be screened for alcohol use disorder using the CAGE questionnaire (need to Cut down, Annoyed by criticism, Guilty about drinking, and need for a morning Eye-opener; see Detecting alcoholism. The CAGE questionnaire) or AUDIT (see Alcohol Use Disorders Identification Test). When the patient's alcohol consumption is in doubt, history can be confirmed by family members or alcohol biomarkers, which include urine or hair ethyl glucuronide, urine ethyl sulfate, and phosphatidylethanol (PEth). PEth is particularly useful because it has a half‐life of approximately 10 to14 days, longer with chronic heavy alcohol consumption.
There is no specific test for alcohol-related liver disease, but if the diagnosis is suspected, tests of hepatocellular injury as well as secretory and synthetic function (prothrombin time [PT]; serum bilirubin, aminotransferase, and albumin levels) and CBC are performed to detect signs of liver injury and anemia.
Elevations of aminotransferases are moderate (< 300 IU/L) and do not reflect the extent of liver damage. The ratio of aspartate aminotransferase (AST) to alanine aminotransferase (ALT) is ≥ 2 (1). The basis for low ALT is a dietary deficiency of pyridoxal phosphate (vitamin B6), which is needed for ALT to function. Its effect on AST is less pronounced. Serum gamma-glutamyl transpeptidase (GGT) increases, more because ethanol induces this enzyme than because patients have cholestasis or liver injury or use other drugs. Serum ). The basis for low ALT is a dietary deficiency of pyridoxal phosphate (vitamin B6), which is needed for ALT to function. Its effect on AST is less pronounced. Serum gamma-glutamyl transpeptidase (GGT) increases, more because ethanol induces this enzyme than because patients have cholestasis or liver injury or use other drugs. Serumalbumin may be low, usually reflecting undernutrition but occasionally reflecting otherwise obvious liver failure with deficient synthesis. Macrocytosis with a mean corpuscular volume > 100 fL reflects the direct effect of alcohol on bone marrow as well as macrocytic anemia resulting from folate deficiency, which is common among people with alcohol use disorder who are undernourished. Indexes of the severity of liver disease are:
Serum bilirubin, which represents secretory function
Prothrombin time or international normalized ratio, which reflects synthetic ability
Thrombocytopenia can result from the direct toxic effects of alcohol on bone marrow or from splenomegaly, which accompanies portal hypertension. Neutrophilic leukocytosis may result from alcoholic hepatitis, although coexisting infection (particularly pneumonia and spontaneous bacterial peritonitis) should also be suspected.
Imaging tests of the liver are not routinely needed for initial diagnosis but can be helpful in evaluating extent of disease, ruling out other disease processes, and monitoring progression (2, 3). Abdominal ultrasound or CT may suggest hepatic steatosis or show splenomegaly, evidence of portal hypertension, or ascites. Vibration-Controlled Transient Elastography (VCTE), ultrasound or magnetic resonance elastrography measures liver stiffness and thus detects advanced fibrosis in the absence of active inflammation. This valuable adjunct can identify degree of fibrosis to assist with future monitoring.
If abnormalities suggest alcohol-related liver disease, screening tests for other treatable forms of liver disease, especially viral hepatitis, should be performed.
Biopsy has a limited role in the diagnosis and prognosis of alcoholic hepatitis and alcohol-related liver disease because biopsy is unlikely to change management which is focused on abstinence and monitoring liver function (3). Proposed indications for liver biopsy include the following:
Unclear clinical diagnosis (eg, equivocal clinical and laboratory findings, unexplained persistent elevations of aminotransferase levels)
Clinical suspicion of > 1 cause of liver disease (eg, alcohol plus viral hepatitis)
Liver biopsy confirms liver disease, helps identify excessive alcohol use as the likely cause, and establishes the stage of liver injury. If iron accumulation is observed, measurement of the iron content and genetic testing can eliminate hereditary hemochromatosis as the cause.
For stable patients with cirrhosis, the American Association for the Study of Liver Diseases (AASLD) recommends that liver ultrasound, with or without alpha-fetoprotein (AFP) measurement, should be performed every 6 months to screen for hepatocellular carcinoma. Because of low anticipated survival for patients with Child's class C cirrhosis who are not on the transplant waiting list, AASLD also suggests surveillance not be performed in these patients (4).
Diagnosis references
1. Kwo PY, Cohen SM, Lim JK. ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries. Am J Gastroenterol. 2017;112(1):18-35. doi:10.1038/ajg.2016.517
2. Maheshwari S, Gu CN, Caserta MP, et al. Imaging of Alcohol-Associated Liver Disease. AJR Am J Roentgenol. 2024;222(1):e2329917. doi:10.2214/AJR.23.29917
3. Crabb DW, Im GY, Szabo G, et al. Diagnosis and Treatment of Alcohol-Associated Liver Diseases: 2019 Practice Guidance From the American Association for the Study of Liver Diseases. Hepatology. 2020;71(1):306-333. doi:10.1002/hep.30866
4. Heimbach JK, Kulik LM, Finn RA, et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology. 67(1):358-380, 2018. doi: 10.1002/hep.29086
Treatment of Alcohol-Related Liver Disease
Abstinence
Supportive care
Corticosteroids and enteral nutrition for severe alcoholic hepatitis
Sometimes transplantation
Restricting alcohol intake
Abstinence is the mainstay of treatment; it prevents further damage from alcohol-related liver disease and thus prolongs life. Because compliance is problematic, a compassionate team approach is essential. Behavioral and psychosocial interventions can help motivated patients; they include rehabilitation programs and support groups (see Alcohol Use Disorders and Rehabilitation: Maintenance), brief interventions by primary care clinicians, and therapies that explore and clarify the motivation to abstain (motivational enhancement therapy).
Medications, if used, should supplement other interventions. Opioid antagonists (naltrexone or nalmefene) and medications that modulate gamma-aminobutyric acid receptors (baclofen or acamprosate) appear to have a short-term benefit by reducing the craving and withdrawal symptoms. Disulfiram inhibits aldehyde dehydrogenase, allowing acetaldehyde to accumulate; thus, drinking alcohol within 12 hours of taking disulfiram causes flushing and has other unpleasant effects. However, evidence for disulfiram is not as strong as for other medications (Medications, if used, should supplement other interventions. Opioid antagonists (naltrexone or nalmefene) and medications that modulate gamma-aminobutyric acid receptors (baclofen or acamprosate) appear to have a short-term benefit by reducing the craving and withdrawal symptoms. Disulfiram inhibits aldehyde dehydrogenase, allowing acetaldehyde to accumulate; thus, drinking alcohol within 12 hours of taking disulfiram causes flushing and has other unpleasant effects. However, evidence for disulfiram is not as strong as for other medications (1, 2).
Supportive care
General management emphasizes supportive care. A nutritious diet and vitamin supplements (especially B vitamins) are important during the first few days of abstinence. Alcohol withdrawal requires use of benzodiazepines (eg, diazepam). In patients with advanced alcohol-related liver disease, excessive sedation can precipitate requires use of benzodiazepines (eg, diazepam). In patients with advanced alcohol-related liver disease, excessive sedation can precipitateportosystemic encephalopathy and thus must be avoided.
Severe acute alcoholic hepatitis commonly requires hospitalization, often in an intensive care unit, to facilitate enteral feeding (which can help manage nutritional deficiencies) and to manage specific complications (eg, infection, bleeding from esophageal varices, specific nutritional deficiencies, Wernicke encephalopathy, Korsakoff psychosis, electrolyte abnormalities, portal hypertension, ascites, portosystemic encephalopathy).
Specific treatment
Corticosteroids (eg, prednisolone 40 mg/day orally for 4 weeks, followed by tapered doses) may improve outcomes in patients who have severe acute alcoholic hepatitis (Maddrey discriminant function Corticosteroids (eg, prednisolone 40 mg/day orally for 4 weeks, followed by tapered doses) may improve outcomes in patients who have severe acute alcoholic hepatitis (Maddrey discriminant function≥ 32 or model for end-stage disease [MELD] score > 20 ) and who do not have infection, gastrointestinal bleeding, renal failure, or pancreatitis (2-4). In a large prospective randomized controlled trial, prednisolone trended toward a decrease in 28-day mortality but did not achieve statistical significance (). In a large prospective randomized controlled trial, prednisolone trended toward a decrease in 28-day mortality but did not achieve statistical significance (5). As a result, corticosteroids may be stopped prior to completing a 4-week course if there is no response to corticosteroids as determined by the day 7 Lille score (6, 7). N-acetylcysteine may be used as an adjunct to corticosteroids (). N-acetylcysteine may be used as an adjunct to corticosteroids (8).
Other than corticosteroids and enteral feeding, few specific treatments are clearly established. Antioxidants (eg, S-adenosyl-L-methionine, phosphatidylcholine, metadoxine) show promise in ameliorating liver injury during early cirrhosis but require further study. Therapies directed at cytokines, particularly tumor necrosis factor (TNF)-alpha, and aiming to reduce inflammation have had mixed results in small trials (9). Despite mixed early results, pentoxifylline, a phosphodiesterase inhibitor that inhibits TNF-alpha synthesis, has not been shown to improve outcomes in patients with severe alcoholic hepatitis (). Despite mixed early results, pentoxifylline, a phosphodiesterase inhibitor that inhibits TNF-alpha synthesis, has not been shown to improve outcomes in patients with severe alcoholic hepatitis (5) . When biologic agents that inhibit TNF-alpha (eg, infliximab, etanercept) are used, risk of infection outweighs benefit. Medications given to decrease fibrosis (eg, colchicine, penicillamine) and medications given to normalize the hypermetabolic state of the alcoholic liver (eg, propylthiouracil) have no proven benefit. Antioxidant remedies, such as silymarin (milk thistle) and vitamins A and E, are ineffective.) . When biologic agents that inhibit TNF-alpha (eg, infliximab, etanercept) are used, risk of infection outweighs benefit. Medications given to decrease fibrosis (eg, colchicine, penicillamine) and medications given to normalize the hypermetabolic state of the alcoholic liver (eg, propylthiouracil) have no proven benefit. Antioxidant remedies, such as silymarin (milk thistle) and vitamins A and E, are ineffective.
Liver transplantation for alcohol-associated cirrhosis should be considered for all patients with decompensated liver disease (ascites, spontaneous bacterial peritonitis, variceal bleeding, hepatic encephalopathy) despite abstinence. With transplantation, 5-year survival rates are comparable to those whose liver disease is not related to alcohol. The American Association for the Study of Liver Diseases (AASLD) and American College of Gastroenterology recognize the limitations of the historical rule requiring 6 months of abstinence from alcohol, instead encouraging candidate evaluation to focus on predicting likelihood of abstinence before and after transplant independent of the length of time of sobriety (4, 10). Clinicians should counsel patients about alcohol misuse treatment programs.
During the COVID-19 pandemic, transplant waitlist registrants increased more than 50% from pre-COVID predictions, possibly due to the increase in alcohol use disorder as well as changes in transplant center risk stratification (11, 12).
Treatment references
1. Perry C, Liberto J, Milliken C, et al. The Management of Substance Use Disorders: Synopsis of the 2021 U.S. Department of Veterans Affairs and U.S. Department of Defense Clinical Practice Guideline. Ann Intern Med. 2022;175(5):720-731. doi:10.7326/M21-4011
2. Crabb DW, Im GY, Szabo G, et al. Diagnosis and Treatment of Alcohol-Associated Liver Diseases: 2019 Practice Guidance From the American Association for the Study of Liver Diseases. Hepatology. 2020;71(1):306-333. doi:10.1002/hep.30866
3. Rambaldi A, Saconato HH, Christensen E, et al. Systematic review: Glucocorticosteroids for alcoholic hepatitis—A Cochrane Hepato-Biliary Group systematic review with meta-analyses and trial sequential analyses of randomized clinical trials. Aliment Pharmacol Ther. 27(12):1167-1178, 2008. doi: 10.1111/j.1365-2036.2008.03685.x
4. Jophlin LL, Singal AK, Bataller R, et al. ACG Clinical Guideline: Alcohol-Associated Liver Disease. Am J Gastroenterol. 2024 Jan 1;119(1):30-54. doi: 10.14309/ajg.0000000000002572. Epub 2023 Sep 1. PMID: 38174913; PMCID: PMC11040545.
5. Thursz MR, Richardson P, Allison M, et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med. 372:1619-1628, 2015. doi: 10.1056/NEJMoa1412278
6. Forrest EH, Atkinson SR, Richardson P, et al. Application of prognostic scores in the STOPAH trial: Discriminant function is no longer the optimal scoring system in alcoholic hepatitis. J Hepatol. 68(3):511-518, 2018. doi: 10.1016/j.jhep.2017.11.017
7. Bataller R, Arab JP, Shah VH. Alcohol-Associated Hepatitis. N Engl J Med. 2022;387(26):2436-2448. doi:10.1056/NEJMra2207599
8. Nguyen-Khac E, Thevenot T, Piquet MA, et al. Glucocorticoids plus N-acetylcysteine in severe alcoholic hepatitis. . Glucocorticoids plus N-acetylcysteine in severe alcoholic hepatitis.N Engl J Med. 2011;365(19):1781-1789. doi:10.1056/NEJMoa1101214
9. Naveau S, Chollet-Martin S, Dharancy S, et al. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. . A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis.Hepatology. 2004;39(5):1390-1397. doi:10.1002/hep.20206
10. Martin P, DiMartini A, Feng S, et al. Evaluation for liver transplantation in adults: 2013 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Hepatology. 2014;59(3):1144-1165. doi:10.1002/hep.26972
11. Anderson MS, Valbuena VSM, Brown CS, et al. Association of COVID-19 With New Waiting List Registrations and Liver Transplantation for Alcoholic Hepatitis in the United States. JAMA Netw Open. 4(10):e2131132, 2021. doi:10.1001/jamanetworkopen.2021.31132
12. Bittermann T, Mahmud N, Abt P. Trends in Liver Transplantation for Acute Alcohol-Associated Hepatitis During the COVID-19 Pandemic in the US. JAMA Netw Open. 4(7):e2118713, 2021. doi:10.1001/jamanetworkopen.2021.18713
Prognosis for Alcohol-Related Liver Disease
Prognosis is determined by the degree of hepatic fibrosis and inflammation. Hepatic steatosis and alcoholic hepatitis without fibrosis are reversible if alcohol is avoided. Fibrosis and cirrhosis are usually irreversible.
Certain biopsy findings (eg, neutrophils, perivenular fibrosis) indicate a worse prognosis. Proposed quantitative indexes to predict severity and mortality use primarily laboratory features of liver failure such as prothrombin time, creatinine (for hepatorenal syndrome), and bilirubin levels. The Maddrey discriminant function may be used; it is calculated from the following formula:

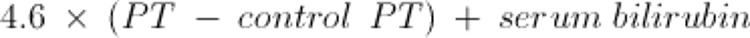

For this formula, bilirubin level is measured in mg/dL (converted from bilirubin in micromol/L by dividing by 17). A value of ≥ 32 is associated with a high short-term mortality rate. Other indexes include the Model for End-Stage Liver Disease (MELD) score, Glasgow alcoholic hepatitis score, and Lille score. For patients ≥ 12 years of age, the MELD score is calculated using the following formula:

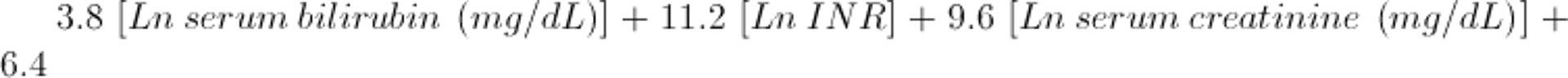
Once cirrhosis and its complications (eg, ascites, bleeding) develop, the overall 5-year survival rate for all patients with alcoholic cirrhosis is approximately 50%, although the survival rate is better for those who do not develop encephalopathy. Long-term abstinence (> 1.5 years) can improve survival (1-3).
Coexisting iron accumulation or chronic hepatitis C increases risk of hepatocellular carcinoma.
Prognosis references
1. Sahlman P, Nissinen M, Pukkala E, et al. Incidence, survival and cause-specific mortality in alcoholic liver disease: a population-based cohort study. Scand J Gastroenterol. 2016;51(8):961-966. doi:10.3109/00365521.2016.1157889
2. Nilsson E, Anderson H, Sargenti K, et al. Incidence, clinical presentation and mortality of liver cirrhosis in Southern Sweden: a 10-year population-based study. Aliment Pharmacol Ther. 2016;43(12):1330-1339. doi:10.1111/apt.13635
3. Xie YD, Feng B, Gao Y, et al. Effect of abstinence from alcohol on survival of patients with alcoholic cirrhosis: A systematic review and meta-analysis. Hepatol Res. 2014;44(4):436-449. doi:10.1111/hepr.12131
Key Points
Risk of alcohol-related liver disease increases markedly in men if they ingest > 60 g alcohol/day (more than 4 drinks per day or 14 drinks per week); risk increases markedly in women if they ingest approximately half (or less) of that amount.
Screen patients using the AUDIT or CAGE questionnaire, and when in doubt about the patient's alcohol consumption, consider use of alcohol biomarkers.
To estimate prognosis, use formulas (eg, Maddrey discriminant function, Model for End-Stage Liver Disease [MELD] score).
Emphasize abstinence, provide supportive care, and hospitalize and consider corticosteroids to patients with severe acute alcoholic hepatitis.
Consider liver transplantation for patients with decompensated liver disease (ascites, spontaneous bacterial peritonitis, variceal bleeding, hepatic encephalopathy) despite abstinence.
Drug Information for the Topic


