



Benign bone tumors include benign giant cell tumors of bone, chondroblastomas, chondromyxoid fibromas, enchondromas, nonossifying fibromas, osteoblastomas, osteochondromas, osteoid osteomas, aneurysmal bone cysts, unicameral bone cysts, and fibrous dysplasia.
Aneurysmal bone cyst
An aneurysmal bone cyst is an expansile lesion that usually develops before age 25 years. This cystic lesion usually occurs in the metaphyseal region of the long bones, but almost any bone may be affected. It tends to grow slowly. A periosteal new bone shell forms around the expansile lesion and is often wider than the original bone. Pain and swelling are common. The lesion may be present for a few weeks to months before diagnosis. In addition to being a de novo benign bone tumor, aneurysmal bone cysts can also occur secondary to other benign bone tumors, typically a giant cell tumor of bone. Primary aneurysmal bone cysts are associated with a rearrangement of the USP6 gene, a finding not present in a secondary aneurysmal bone cyst.
The appearance on radiograph is often characteristic: The lucent area is usually well circumscribed and eccentric; the periosteum bulges (balloons) and extends into the soft tissues and may be surrounded by new bone formation. MRI typically shows fluid-fluid levels on axial and sagittal reconstructions. On imaging, some aneurysmal bone cyst–like lesions may appear more ominous, having characteristics similar to those of osteosarcoma, and thus should raise suspicion of telangiectatic osteosarcoma. A solid variant aneurysmal bone cyst can be confused radiographically for a giant cell tumor of bone at the very end of the bone.
A biopsy-confirmed aneurysmal bone cyst warrants treatment, as these can behave aggressively and cause local destruction. Contained lesions can be injected with a mixture of doxycycline, albumin, and air that forms an injectable foam (1). The goal is to inject each portion of the cyst, and in many occasions more than one injection may be needed. Other sclerosing alcohol-based agents have been used. Surgical removal of the entire lesion is another effective treatment. Radiation may be the treatment of choice in completely surgically inaccessible vertebral lesions that are compressing the spinal cord. However, radiation is avoided whenever possible due to the risk of developing secondary malignancies.

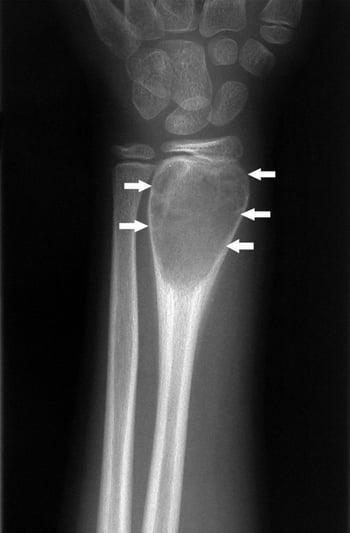
This wrist radiograph shows periosteal new bone formation around the expansile lesion (arrows), which is characteristic of an aneurysmal bone cyst.
Benign giant cell tumor of bone
Benign giant cell tumors of bone, which most commonly affect people in their 20s and 30s, occur in the epiphyseal and distal metaphyseal-epiphyseal areas. These tumors are considered to be very locally aggressive. They continue to enlarge and destroy bone and may eventually erode and extend into the soft tissues. They may cause pain. These tumors are notorious for their tendency to recur. Rarely, a giant cell tumor of bone may metastasize to the lung, even though it remains histologically benign. The prevalence of these benign pulmonary metastases is approximately 4 to 7% of patients (2). Lesions that are more locally destructive, have recurred, and present in axial locations are more prone to metastasize.
Benign giant cell tumors of bone appear as expansile lytic lesions on imaging. On imaging studies, there is a margin without a sclerotic rim where the tumor ends and normal trabecular bone begins. Biopsy is necessary to confirm the diagnosis. Because of the potential for pulmonary metastases, a chest CT is done as part of initial staging and chest radiographs or CT are performed for routine surveillance after treatment.
Most benign giant cell tumors of bone are treated by radical curettage, local chemical or thermal adjuvants, and packing with methyl methacrylate or by bone graft. To reduce recurrence rate, surgeons often prefer using an adjuvant, such as thermal heat delivered by argon beam, freezing with liquid nitrogen, or treating the tumors chemically with phenol or hydrogen peroxide. If a tumor is very large and destructive to the joint, complete excision with joint reconstruction may be necessary. The monoclonal antibody denosumab, a receptor activator of nuclear factor kappa-B ligand (RANKL) inhibitor, can be used in the treatment of benign giant cell tumor of bone for tumors not amenable to complete resection (see image Giant Cell Tumor [CT Scan]) or for the treatment of pulmonary metastases.


This radiograph of the knee shows a radiolucent lesion in the lateral distal femur above the knee, which is characteristic of a giant cell tumor of bone.

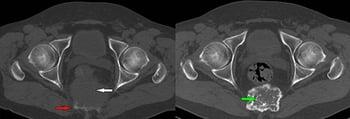
This axial CT scan (left) shows a lytic lesion of the sacrum (bottom blue arrow) with anterior soft tissue mass (top white arrow). A biopsy confirmed giant cell tumor of bone. Following denosumab treatment, the follow-up CT scan (right) shows progressive ossification of the tumor (bottom black arrow) without an increase in size.
Chondroblastoma
Chondroblastoma is rare and occurs most commonly in individuals less than 20 years old. Arising in the epiphysis, this tumor may be locally aggressive, causing destruction to the bone and the joint.
Chondroblastoma appears on imaging studies as a sclerotic marginated radiolucent lesion in an epiphyseal or apophyseal location and possesses intralesional calcification. MRI can help diagnostically by showing significant edema around the lesion and CT can be helpful to better demonstrate the calcifications if they are not apparent on radiographs.
This neoplasm is typically treated with aggressive curettage, and the cavity is bone grafted with allograft bone or bone substitute. Local recurrence rate is in the range of 5 to 10% when treated with surgical curettage (3, 4), and recurrent lesions often resolve with repeat bone curettage and bone grafting. In some instances, prophylactic internal fixation may be warranted if the curettage has created a substantial cortical defect.

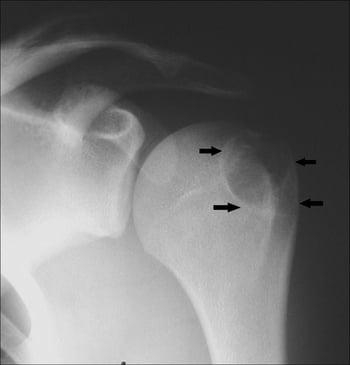
This shoulder radiograph shows a chondroblastoma in the humerus at the greater tuberosity apophysis (arrows).
Chondromyxoid fibroma
Chondromyxoid fibroma is very rare and usually occurs in younger patients, typically those less than 30 years old.
The appearance of a chondromyxoid fibroma on imaging studies is an eccentric, sharply demarcated lytic lesion located near the end of long bones. The proximal tibia and iliac wing are common locations.
Treatment of chondromyxoid fibroma after biopsy is surgical excision or curettage, often use of an adjuvant (eg, phenol, liquid nitrogen, use of an argon beam), and bone grafting. These are typically treated very aggressively due to their locally destructive nature and propensity to recur if incompletely treated.

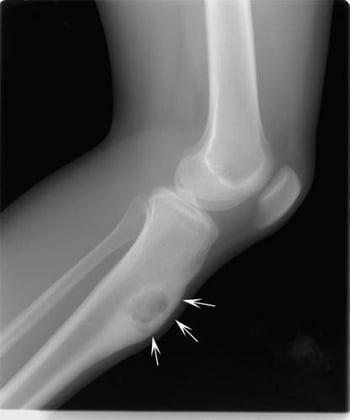
This radiograph of the knee shows a chondromyxoid fibroma in the tibia below the knee (arrows), expanding the bone in the front.
Enchondroma
Enchondromas may occur at any age but tend to manifest in adulthood. They are usually located within the medullary bone in the metaphyseal-diaphyseal region. These tumors are usually asymptomatic and detected incidentally (eg, in imaging to evaluate a musculoskeletal injury). However, on occasion enchondromas can be large and rarely can cause pain.
On radiograph, the lesion may appear as a lobulated calcified area within bone; some lesions are less calcified, with areas of stippled calcification on either plain films or CT. If adjacent to the cortex, enchondromas often show minor endosteal scalloping. Significant endosteal scalloping should raise concern of a chondrosarcoma. Almost all enchondromas have increased uptake on a bone scan and have the potential to create false concern of cancer. Radiographic findings, including MRI and CT, may be diagnostic; if they are not, and especially if the tumor (not the associated joint) is painful, the diagnosis of enchondroma should be questioned. To help differentiate bone pain from joint pain, the joint can be injected, usually with a long-lasting anesthetic (eg, bupivacaine); if pain persists following the injection, it may be caused by the bone lesion. Biopsy can be considered, but it is extremely difficult to differentiate benign and low-grade malignant cartilage on biopsy (5). The diagnosis of an enchondroma versus a low-grade chondrosarcoma is best made on clinical and radiographic criteria.
An asymptomatic enchondroma does not need biopsy, excision, or other treatment; however, follow-up imaging studies are indicated to rule out the rare disease progression to chondrosarcoma. These studies are typically done at 6 months and again at 1 to 2 years. The patient should be counseled to return any time new symptoms develop, specifically development of worsening pain not relieved by NSAIDs or rest or development of a mass.
Patients with multiple enchondromas (Ollier disease) and especially multiple enchondromatosis with soft tissue hemangiomas (Maffucci syndrome) have a higher risk of chondrosarcoma and development of other malignancies. As such, they warrant closer follow-up.

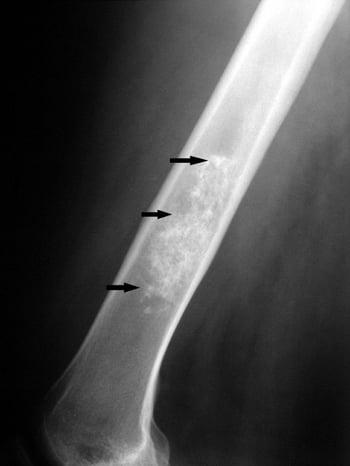
This radiograph of the femur shows lobulated matrix calcifications within the medullary cavity (arrows), which are typical of enchondroma.
Fibrous dysplasia
Fibrous dysplasia involves abnormal bone development during childhood. It weakens bone, which typically results in formation of stress fractures which, in turn, cause slow remodeling and deformation of the bone. Fibrous dysplasia may affect one (monostotic) or several (polyostotic) bones. Polyostotic fibrous dysplasia, cutaneous pigmentation in the form of café-au-lait spots, and endocrine abnormalities resulting in precocious puberty may be present (also known as McCune-Albright syndrome). The abnormal bone lesions of fibrous dysplasia commonly stop developing at puberty. Malignant degeneration is an extremely rare phenomenon.
On radiograph, the lesions can appear cystic and may be extensive and deforming. On imaging, the lesions have a classic ground-glass appearance. Proximal femur lesions, if large enough and left untreated may result in the development of a "shepherd's crook" deformity on radiograph.
Bisphosphonates have been shown to be helpful to relieve pain (6). Progressive deformities, fractures that do not heal with immobilization, or intractable pain may be effectively treated surgically.

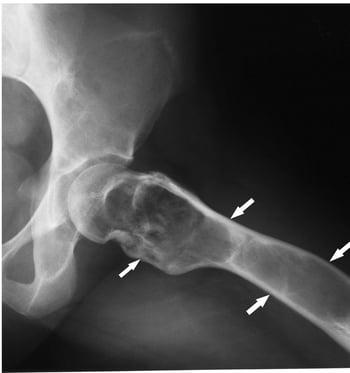
This radiograph of the hip shows fibrous dysplasia (arrows) with cortical expansion and ground-glass matrix.
Melorheostosis
Melorheostosis is a rare developmental disease of cortical bone. It is a mesodermal dysplasia of the cortex that may be linked to a random MAP2K1 gene mutation. The process starts in childhood with thickening of the bone cortex on both the outer surface (periosteal) and inner surface (endosteal). The process evolves very slowly in the cortical bone. Melorheostosis usually affects only a single bone (monostotic) but may affect other bones (polyostotic) of the same limb (somatic distribution). The tibia and femur are the most common sites.
Patients often present with bone pain or discomfort, and radiographs demonstrate thickening of the cortex of the involved bone. The radiographic appearance may suggest traumatic new bone formation, a stress fracture, or even an malignant bone-forming neoplasm (eg, osteosarcoma). However, the dense cortical thickening has a characteristic “dripping candle wax” appearance (irregular cortical hyperostosis), and image stability on serial radiographs is sufficient to make the diagnosis without biopsy.
The patient's symptoms of pain and/or discomfort are best treated with analgesics or anti-inflammatory agents, and serial radiographs are recommended over time. There are also multiple reports of the pain being improved with infusion of bisphosphonates (6). Attempts at surgical removal or en bloc resection for severely painful lesions have usually not been successful, and on very rare occasions, amputation is needed.

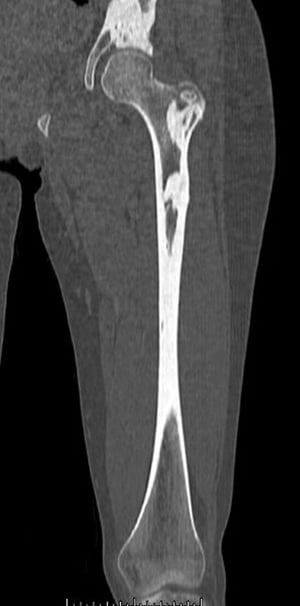
This CT scan shows the dense bone lesions of melorheostosis in the femur and pelvis.
This CT scan shows the dense bone lesions of melorheostosis in the femur and pelvis.
Image courtesy of Michael J. Joyce, MD, and David M. Joyce, MD.

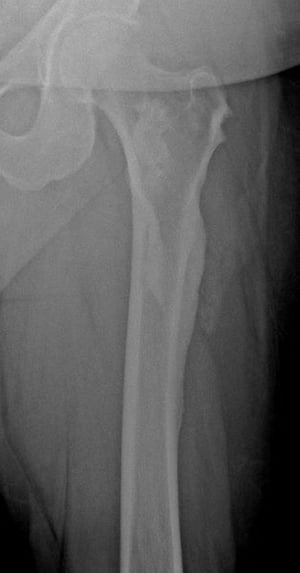
This radiograph shows the "dripping candle wax" appearance of melorheostosis in the femur; it is particularly apparent at the lateral femoral cortex.
This radiograph shows the "dripping candle wax" appearance of melorheostosis in the femur; it is particularly apparent
Image courtesy of Michael J. Joyce, MD, and David M. Joyce, MD.
This CT scan shows the dense bone lesions of melorheostosis in the femur and pelvis.
This CT scan shows the dense bone lesions of melorheostosis in the femur and pelvis.
Image courtesy of Michael J. Joyce, MD, and David M. Joyce, MD.
This radiograph shows the "dripping candle wax" appearance of melorheostosis in the femur; it is particularly apparent at the lateral femoral cortex.
This radiograph shows the "dripping candle wax" appearance of melorheostosis in the femur; it is particularly apparent
Image courtesy of Michael J. Joyce, MD, and David M. Joyce, MD.
Nonossifying fibroma (fibrous cortical defect, fibroxanthoma)
Nonossifying fibroma is a benign fibrous lesion of bone that appears as a well-defined eccentric lesion on radiograph. A nonossifying fibroma involving the periosteal cortical surface is called a fibrous cortical defect. These lesions are essentially developmental defects in which parts of bone that normally ossify are instead replaced with a focal area of fibrous tissue. It appears to be driven by an activating mutation in MAP-kinase signaling (8). These lesions most commonly affect the metaphyses, and the most commonly affected sites are the distal femur, distal tibia, and proximal tibia. They can progressively enlarge and become multiloculated. Nonossifying fibromas are common among children. Most lesions eventually ossify and undergo remodeling, often resulting in dense, sclerotic areas. However, some lesions persist or enlarge and can cause pain related to stress fractures or even outright pathologic fractures.
Most nonossifying fibromas are small and asymptomatic. However, lesions that involve approximately 50% or more of the bone diameter tend to cause pain and increase the risk of pathologic fracture.
Nonossifying fibromas are generally first noted incidentally on imaging studies (eg, after minor injury, sports injury or trauma). They typically are radiolucent and solitary with an oblong lucent appearance and geographic border in the cortex. They can also be multiloculated. There is a syndrome where patients can have multiple nonossifying fibromas along with café-au-lait skin lesions, known as Jaffe-Campanacci syndrome (9).
Small nonossifying fibromas require no treatment and limited follow-up. Lesions that may be causing pain or are close to 50% of the bone diameter may warrant curettage and bone grafting to decrease risk of a pathologic fracture through the lesion.

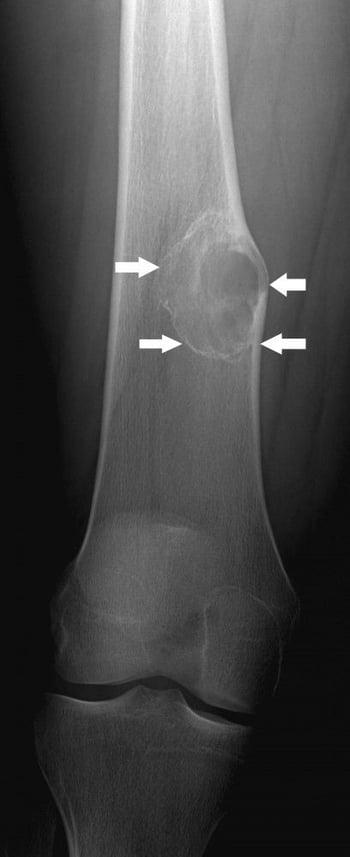
This radiograph shows a bone lesion (arrows) above the knee in the femur with mild expansion, which is typical of nonossifying fibroma.
Osteoid osteoma
Osteoid osteoma, a benign bone tumor that tends to affect young patients (commonly aged 5 to 25), can occur in any bone but is most common in long bones. It can cause pain that is usually worse at night, reflecting increased nocturnal prostaglandin-mediated inflammation. Pain is characteristically relieved by nonsteroidal anti-inflammatory drugs (NSAIDs) that target prostaglandins. In growing children, the inflammatory response and associated hyperemia, if close to the open growth plate, may cause overgrowth and limb length discrepancy. Physical examination may reveal atrophy of regional muscles because the pain causes muscle disuse. In the spine, the inflammation from the lesion can cause muscular spasm and a presentation similar to that of scoliosis, with associated pain.
Characteristic appearance on imaging studies is a small radiolucent nidus surrounded by a sclerotic zone. If the diagnosis is unclear, a total body technetium-99m bone scan will demonstrate the osteoid osteoma as an area of increased uptake. Radiographs or thin-cut CT without contrast are the imaging modalities of choice for establishing the diagnosis, as they will demonstrate the classic "bull's eye" appearance of the true nidus of the tumor surrounded by reactive bone. MRI may also be able to do so, but often with less clarity. MRI will demonstrate a large surrounding area of edema on fluid-sensitive sequences (T2 or STIR), which is representative of the associated inflammation. History and imaging are sufficient for making the diagnosis of osteoid osteoma; however, the small bones of the hands and feet may have a more aggressive appearance, elevating the role of biopsy in those anatomic locations.
Ablation of the small radiolucent zone with percutaneous radiofrequency ablation (RFA) provides permanent relief in most cases (10, 11). As such, this is often treated by an interventional musculoskeletal radiologist. In some instances osteoid osteomas may require surgical curettage. This is most often in the event that the osteoid osteoma is near a critical neurovascular structure or close to the skin (eg, spine, hands, feet) because the heat produced by radiofrequency ablation may cause thermal damage.

Axial CT image of the proximal femur (adjacent bone is pelvic ramus) demonstrating the typical radiolucent nidus with central calcification (arrow) and surrounding reactive rim in a child with left hip pain.
Axial CT image of the proximal femur (adjacent bone is pelvic ramus) demonstrating the typical radiolucent nidus with c
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.

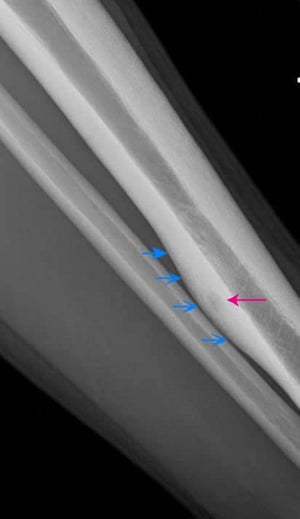
This lateral radiograph of the left tibia shows a lucent posterior cortical osteoid osteoma (red arrow). Note the chronic-appearing, smooth periosteal thickening around the tumor (blue arrows).
This lateral radiograph of the left tibia shows a lucent posterior cortical osteoid osteoma (red arrow). Note the chron
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
Axial CT image of the proximal femur (adjacent bone is pelvic ramus) demonstrating the typical radiolucent nidus with central calcification (arrow) and surrounding reactive rim in a child with left hip pain.
Axial CT image of the proximal femur (adjacent bone is pelvic ramus) demonstrating the typical radiolucent nidus with c
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
This lateral radiograph of the left tibia shows a lucent posterior cortical osteoid osteoma (red arrow). Note the chronic-appearing, smooth periosteal thickening around the tumor (blue arrows).
This lateral radiograph of the left tibia shows a lucent posterior cortical osteoid osteoma (red arrow). Note the chron
Image courtesy of Michael J. Joyce, MD, and Hakan Ilaslan, MD.
Osteoblastoma
Osteoblastoma is a rare benign tumor that consists of tissue histologically identical to that of an osteoid osteoma. Some experts simply consider them large osteoid osteomas (> 2 cm). Osteoblastoma is more common among males and appears typically between ages 10 and 35. The tumor develops in the bone of the spine (posterior elements), legs, hands, and feet. It is a slow-growing tumor that is locally destructive to normal bone and can cause significant pain.
Imaging with radiographs, CT, and MRI will demonstrate a large osteoblastic (bone-forming) mass, which is typically expanded beyond the normal cortical boundaries. Usually an open biopsy is warranted to confirm the diagnosis of osteoblastoma and exclude malignant etiologies.
Treatment of osteoblastoma requires surgery, often curettage and bone grafting. The local recurrence rate for lesions that are treated with intralesional curettage may be as high as 10 to 20%. More aggressive-appearing lesions are treated with surgical en bloc resection and bone reconstruction with either allograft or prostheses. A variation called aggressive osteoblastoma is similar to osteosarcoma both radiographically and histologically.
Osteochondroma
Osteochondromas (osteocartilaginous exostoses), the most common benign bone tumor, may arise from any bone but tend to occur near the ends of long bones. These tumors manifest most often in people aged 10 to 20 and may be single or multiple. Multiple osteochondromas are typically part of a syndrome known as multiple hereditary exostoses (MHE), which is a result of an autosomal dominant mutation in the EXT1, EXT2, or EXT3 genes (12). Malignant degeneration into a secondary chondrosarcoma develops in well under 1% of patients with single osteochondromas, and is almost always a low-grade tumor. Patients with MHE have more tumors and are more likely to develop a chondrosarcoma than patients with a single osteochondroma.
On imaging studies, the lesion appears as a bony prominence with a cartilage cap (usually < 2 cm) off the surface of the bone with no underlying cortex under the prominence. MRI may be helpful in differentiating among a thick cartilage cap, bursa, or surrounding soft tissue mass. The main diagnostic feature of an osteochondroma on imaging is that the medullary canal of the bone is in continuity with the medullary canal associated with the base of the exostosis. Occasionally, a painful bursa may form over the cartilage cap because these lesions are most often periarticular in areas associated with a significant amount of soft tissue motion. Another potential reason for pain in one of these lesion is fracture of the lesion at its base.
Excision is indicated if the tumor is enlarging, causes pain due to the inflammatory bursa, disturbs growth, or demonstrates imaging features concerning for a malignant transformation such as a destructive appearance, soft tissue mass, or thickened cartilaginous cap (> 2 cm). An enlarging tumor or development of unexplained new pain in a known tumor in an adult should raise concern of chondrosarcoma and the possible need for excision or biopsy.

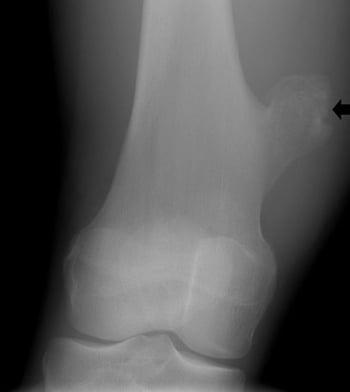
This radiograph of the knee shows a bony outgrowth (arrow) from the femur above the knee joint that is consistent with an osteochondroma.
Enostosis (Bone Island)
Enostoses (bone islands) are small osteomas best thought of as a dense island of cortical bone within the medullary canal. Typical osteomas are benign bone-forming tumors that are uniformly dense and most common in the skull, rarely occurring in the axial skeleton or pelvis. These are not painful and often noted incidentally as bone densities with distinct borders on radiographs. Diagnosis is based on radiographic appearance. On nuclear bone scan, the lesions show little or no increased uptake. If the diagnosis is questionable, MRI imaging can help as the mass/lesion should be uniformly low on T1- and T2-weighted imaging and with no evidence of contrast enhancement. Enostoses and osteomas do not require biopsy and can be followed periodically with plain films.
Osteopoikilosis
Osteopoikilosis is a benign sclerosing dysplastic bone disease with autosomal dominant inheritance characterized by numerous bone islands throughout the skeleton. The lesions are often too numerous to count. It usually involves the axial skeleton and ribs but sometimes involves the extremities, particularly the proximal femurs. Osteopoikilosis causes no symptoms.
Osteopoikilosis is usually encountered incidentally on imaging done for another reason. Diagnosis of osteopoikilosis is based on radiographic appearance; and on serial follow-up, lesions should retain the same appearance. These bone islands show little or no activity on nuclear bone scan. In adults over 40 years old, metastatic carcinoma must be considered in the differential diagnosis. However, metastatic lesions are usually larger, have more irregular margins, and have areas of enhancement on MRI imaging, whereas osteopoikilosis lesions are more numerous in periarticular areas.

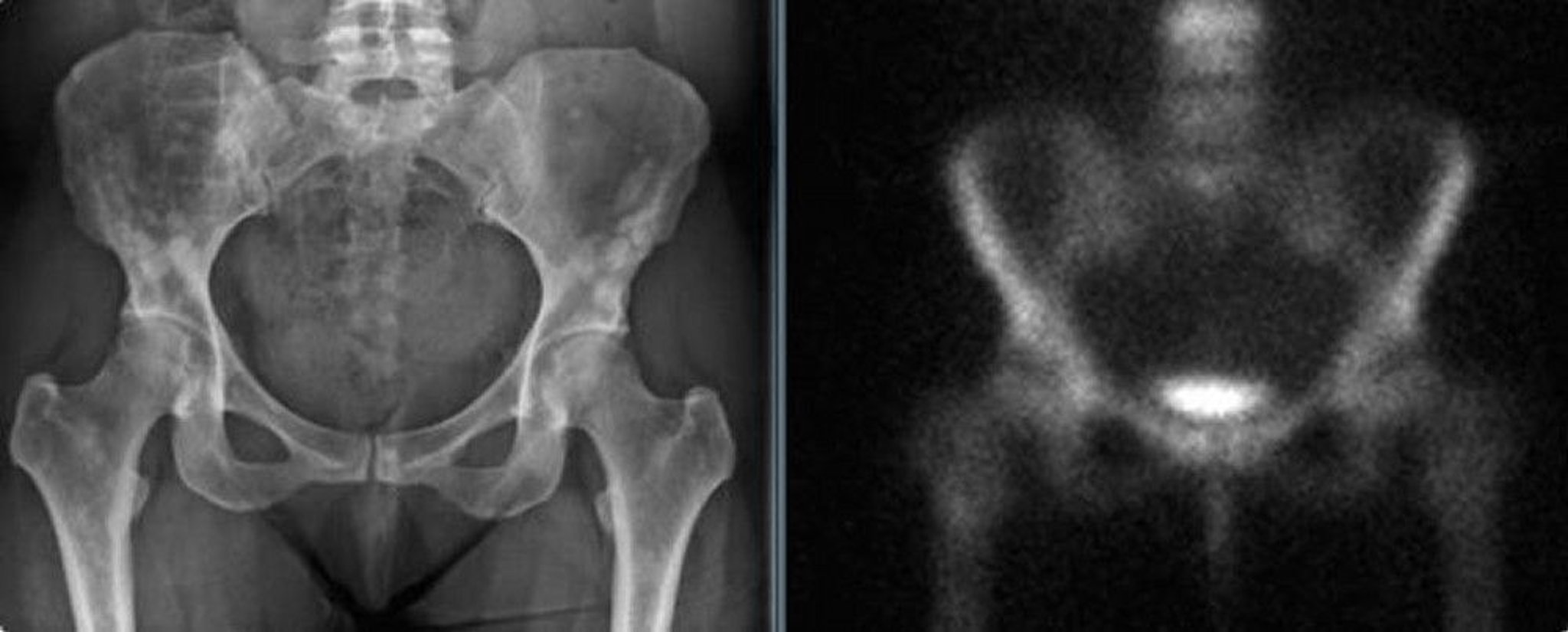
The pelvic radiograph (left) shows osteopoikilosis of the pelvis in an adult woman. These densities could be mistaken for blastic metastases. The bone scan (right) shows no significant increased uptake.
Images courtesy of Michael J. Joyce, MD, and David M. Joyce, MD.
Unicameral bone cyst
Unicameral (or simple) bone cysts occur in the long bones starting distal to the physeal plate in children. The cyst is fluid-filled. It causes the cortex to thin and predisposes the area to a buckle-like pathologic fracture causing intense pain, which is usually how the cyst is recognized.
Radiographs are usually diagnostic. Unicameral bone cysts typically appear as well-marginated lesions without reactive sclerosis or an expansile cortex. If the cyst has a minor fracture, a bone fragment from the thin shell may fall to the bottom of the fluid-filled cyst. The result is the classic "fallen leaf" sign on radiograph.
Smaller cysts sometimes heal without treatment. A nondisplaced fracture through small cysts may be a stimulus for healing; however, that is unpredictable. Larger cysts, particularly in children, may require intervention. Options for treatment include percutaneous injections of corticosteroids, demineralized bone matrix or synthetic bone substitutes, or open curettage and grafting. It is felt that puncturing the cyst wall on the intramedullary side to allow for inflow of bone marrow elements is helpful (13). The response to corticosteroid injections may be variable and may require multiple injections.

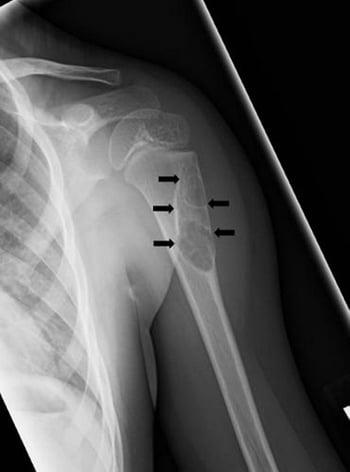
This radiograph shows a cystic lesion (arrows) of the humerus in child consistent with a unicameral bone cyst.
References
1. Shiels WE 2nd, Mayerson JL. Percutaneous doxycycline treatment of aneurysmal bone cysts with low recurrence rate: a preliminary report. Clin Orthop Relat Res. 2013;471(8):2675-2683. doi:10.1007/s11999-013-3043-2
2. Chan CM, Adler Z, Reith JD, Gibbs CP Jr. Risk factors for pulmonary metastases from giant cell tumor of bone. J Bone Joint Surg Am. 2015;97(5):420-428. doi:10.2106/JBJS.N.00678
3. Xu H, Nugent D, Monforte HL, et al. Chondroblastoma of bone in the extremities: a multicenter retrospective study. J Bone Joint Surg Am. 2015;97(11):925-931. doi:10.2106/JBJS.N.00992
4. Huang C, Lü XM, Fu G, Yang Z. Chondroblastoma in the Children Treated with Intralesional Curettage and Bone Grafting: Outcomes and Risk Factors for Local Recurrence. Orthop Surg. 2021 Oct;13(7):2102-2110. doi: 10.1111/os.13153. Epub 2021 Oct 2. PMID: 34599644; PMCID: PMC8528993.
5. Skeletal Lesions Interobserver Correlation among Expert Diagnosticians (SLICED) Study Group. Reliability of histopathologic and radiologic grading of cartilaginous neoplasms in long bones. J Bone Joint Surg Am. 2007;89(10):2113-2123. doi:10.2106/JBJS.F.01530
6. Chapurlat R, Legrand MA. Bisphosphonates for the treatment of fibrous dysplasia of bone. Bone. 2021;143:115784. doi:10.1016/j.bone.2020.115784
7. Hollick RJ, Black A, Reid D. Melorheostosis and its treatment with intravenous zoledronic acid. BMJ Case Rep. 2010;2010:bcr04.2009.1757. doi:10.1136/bcr.04.2009.1757
8. Baumhoer D, Kovac M, Sperveslage J, et al. Activating mutations in the MAP-kinase pathway define non-ossifying fibroma of bone. J Pathol. 2019;248(1):116-122. doi:10.1002/path.5216
9. Sabry AO, Abolenain AS, Mostafa N, Ramadan A, Ghanem M. Jaffe-Campanacci syndrome; a case series and review of the literature. BMC Musculoskelet Disord. 2024;25(1):502. Published 2024 Jun 27. doi:10.1186/s12891-024-07581-0
10. Cerny J, Soukup J, Cerna S, Novotny T. Current Approaches to Osteoid Osteoma and Minimally Invasive Surgery-A Minireview and a Case Report. J Clin Med. 2022;11(19):5806. Published 2022 Sep 30. doi:10.3390/jcm11195806
11. Rehnitz C, Sprengel SD, Lehner B, Ludwig K, Omlor G, Merle C, Kauczor HU, Ewerbeck V, Weber MA. CT-guided radiofrequency ablation of osteoid osteoma and osteoblastoma: clinical success and long-term follow up in 77 patients. Eur J Radiol. 2012 Nov;81(11):3426-34. doi: 10.1016/j.ejrad.2012.04.037. Epub 2012 Jul 6. PMID: 22770580.
12. Bukowska-Olech E, Trzebiatowska W, Czech W, et al. Hereditary Multiple Exostoses-A Review of the Molecular Background, Diagnostics, and Potential Therapeutic Strategies. Front Genet. 2021;12:759129. Published 2021 Dec 10. doi:10.3389/fgene.2021.759129
13. Pretell-Mazzini J, Murphy RF, Kushare I, Dormans JP. Unicameral bone cysts: general characteristics and management controversies. J Am Acad Orthop Surg. 2014;22(5):295-303. doi:10.5435/JAAOS-22-05-295
Drug Information for the Topic





